0.0张和液体的透过性最小:聚丙烯可以在开水中煮,在135℃、100h的蒸汽中消毒也不
聚合物基复合材料第5章环氧树脂 (3)改性多元服 由于当使用政性多元度对人的应装和基服有刺激性,与环意的(2)咪唑类固化剂 咪唑类化合物
聚合物基复合材料
第5章
环氧树脂 (3)改性多元服 由于当使用政性多元度对人的应装和基服有刺激性,与环意的
(2)咪唑类固化剂 咪唑类化合物是一种新型固化剂,可在较低温度下固化而得到耐热性的,被往请加成多元族将过量的多元成与单环氧化合物或双环氧化合物反应而得
优良的固化物,并且具有优异的力学性能。
改性多元胺,生成物通常为胺加成物:
表5—14 具有代表性的叔胺类固化剂RNH:+CH:-CH-R'→RNHCH:-CH2-R'
名称 略称
化学结构 0
OH
直链二胺 (CH3)2N(CH2)。N(CH1):
因为加成物分子量增大,沸点和黏度增高,对人的皮肤和粘膜的刺激性随之大幅度减小,直链叔胺
(CH1)2N(CH2)_CH 同时由于加成反应生成羟基,提高了圆化反应活性。有代表性的这类加成物是DETA与米基薯
NH 水甘油醚或与低分子量的DGEBA树脂的加成物。
脂肪胺 四甲基胍
TMG
(CH3):NCN(CH3): ②迈克尔加成多元胺胺的活泼领对a、β不饱和链能迅速加成反应,称为迈克尔加成反
叔烷基单胺 N[(CH2).CH3],
点。此反应是在氨基上进行的加成反应,因此改善了改性多元胺的刺激性和对环氧树脂的相三乙醇胺
TEA N(CH2CH,OH),
性,特别是丙烯腈的加成反应称为腈乙基化,在延缓反应活性和改善相容性方面是非常有效的,RNH:+CH:-CH-C=N→RNHCH2-CH:CN
HN CH:
CH:-CH:
哌啶 ③曼尼斯加成多元胺 曼尼斯反应为多元胺、福尔马林以及苯酚的缩合反应。此反应可大
CH:-CH: 幅度改善固化特性,能够低温固化。这种改性固化剂的性质,根据胺和酚的种类以及它们的配
CH:-CH: 比不同而不同。
脂环胺 N.N'—二甲基哌嗪
CHaN NCH
OH
CH:-CH: HO
RNH:+HCHO+ RNHCH:
+H20 CH2-CH
5.3.3.2 叔胺及咪唑类固化剂
CH2-CH: 三亚乙基二胺
-CH2-CH2
(1)叔胺类固化剂 叔胺属于碱性化合物,是阴离子型的催化型固化剂。它与环氧树脂的固化反应机理如下:
吡啶 Pyr
R2N+-CH:-CH-CH2~→R,N-CH2-CH-~
0
e0
杂环胺 甲基吡啶
MPyr N CH3
R,NO-CH2-CH-~+n(CH2-CH-~)→R2N(CH1-CH-O),CH2-CH-~
0
1.8—二氮双环(5,4,
DBU 60
80
0)-7-十一烯 叔胺类固化剂具有固化剂用量、固化速度和
CH2N(CH3)2 固化物性能变化较大,固化时放热较大的缺点,
苄基二甲胺 BDMA
100 因此不适应于大型浇注,也不应单独使用。表5—14
列出了具有代表性的叔胺类固化剂。
CH:N(CH3)2
OH 90
DMP-10 表5—14给出的叔胺类固化剂是属于阴离子聚
苯酚 2—(二甲氨基甲基)
合催化型的叔胺化合物。用叔胺类化合物作为固
80
OH 化剂固化的DGEBA树脂的热变形温度(HDT)
(CH3)2NH2C CH2N(CH3)z
如图5—3所示,由图可看出不同胺类固化剂在不同
芳香胺
2.4.5—三(二甲氨基70
温度下进行固化,其固化物的HDT也不同,对固
化温度的影响是很显著的。即使同一种固化剂在甲基)苯酚
DMP-30
60 1
固化温度90℃时,所有叔胺类固化环氧树脂的不同温度下固化,其固化物的HDT也相差较大。
60 90 120 150
180 CH:N(CH3):
固化温度/℃
OH CH2CH
图5—3 固化温度对 HDT的影响
(CH3):NH2C
HDT值最大;如果固化温度超过这一温度,HDT
DMP—30的三—2—乙
CH2N(CH3)2 固化剂添加量5%(摩尔分数),固化时间30h
反而下降。叔胺类固化剂固化环氧树脂的固化物,
基己酸盐
CH2N(CH3)2 3CH,CH2CH2CH2CHCOOH
1—N.N—二甲基正已胺;2—N.N—二甲基环己基胺;
氧树脂固化物的HDT相比是非常低的。这可能是其HDT与咪唑类化合物或三氟化硼配合物固化环
3—N.N—二甲氨基甲基甲酚;4—N,N—二甲基苄胺:
5—N,N—二甲氨基甲基苯酚
叔胺的分解使链增长受阻之故。074
075 HDT/C
聚合物基复合材料
第5章 环氧树脂 味啡类化合物的反应活性根据其结构不同面有所不同,一般来说,碱性意强因化温度
这是因为1位上的氢与3位上的氮不能共振所致。
因化解程是具有的便化作用,又有仲废的作用,味理的碱性比较弱且挥发性小,请性也就比为了位上的个原子的五元环,一个氮原子构成神肢,一个氯原子构成叔除,所以降重
180 160
凝胶化后在各后固化温度下固化4h
的族胺、芳香族胺小得多,通常它们在250℃以下几乎不分解。各种咪唑的特性见表5—15,
表5—15 有代表性的咪唑化合物的结构与特性编号
凝胶化温度/C 后固化温度/℃
沸点/℃ 活性温度/℃
物
态 120
1
30 93
名称及结构式 熔点/℃
2
30 149
7 3
30 204
2-甲基咪唑 10
CH-N 263~265
白色结晶 2—乙基—4—甲基咪唑添加量/质量份
82~87 1
CH C-CH 135~139
(b)
N 图5—4 2—乙基—4—甲基咪唑的用量固化条件对热变形温度的影响
H
2—乙基—4—甲基咪唑
CH3-C-N 150g
45 292
82~87 淡黄色黏性液体
CH
C-C:H 140
H 130
2-乙基味唑
CH-N 120
1 61~66
270~275 82~87
白色结晶 CH
C-C:Hs 110
N 1
2 3 4
H 时间/d
2.4-二甲基咪唑 图5—5 2—乙基—4—甲基味唑与间苯二胺的热老化比较
CH1-C-
N 环氧树脂的环氧当量190,在204℃老化4天
H 92
276~278 白色结晶
1—14%(质量分数)间苯二胺:2—2.9%(质量分数)2—乙基—4—甲基咪唑82~87
も
H 5.3.3.3 酸酐类固化剂
环氧树脂和多元酸反应速率很慢,由于不能生成高交联度产物,因而不能作为固化剂之用。
咪唑类化合物中最引人注目的是2—乙基—4—甲基咪唑。在室温下易与环氧树脂混合,得到低酸酐由于具有使用寿命长、对皮肤基本上没有刺激性、固化反应缓慢、放热量小、收缩率低、
黏度混合物,适用期长。固化物有较高的热变形温度,当用量为2%时,经150℃/4h固化后马产物的耐热性高、产物的机械强度、电性能优良等优点而成为一类重要的固化剂。
丁耐热温度达160℃。如果在树脂混合物中添加125%的二氧化硅填料,固化物的机械强度有明酸酐和环氧树脂的反应机理与其有无促进剂存在而有所不同。
显提高,马丁耐热温度大于225℃。由图5—4和图5—5可见,2—乙基—4—甲基咪唑的树脂固化物和①无促进剂存在时
酸酐及芳香胺的比较,经高温、短时间的后固化处理后可得到相当高的热变形温度(约240℃),a.首先由环氧树脂的羟基与酸酐反应生成含酯链的羧酸:
这种特性对制备缠绕结构的复合材料是极其有利的。
160 凝胶化后在各后固化温度下固化4h
R 0+HO-
R
140 编号
凝胶化温度/℃ C
C-O 后固化温度/℃
1
1
70 0
0
120 70
2
70 93
b.羧酸和环氧树脂的环氧基开环加成反应生成仲羟基:
3
70 149
0 100
4
70 10
204
2—乙基4—甲基咪唑添加量/质量份R
+CH2-CH (a)
C-OH C-O-CH2-CH
0 HO
076 077
热变形温度/℃ 热变形温度/℃
热变形温度/℃
聚合物基复合材料
第5章 环氧树脂
c.生成的羟基和另一个酸酐反应(反应式同1a.),与上述反应同时进行的是反应d.。
在无促进剂存在时K值为0.8~0.9,在有促进剂存在时K值为1.
L0
酸酐固化剂可分为单一型、混合型、共熔混合型,其中单一型又分为单官能团酸酐、双官d.生成的仲羟基再和另一个环氧基反应:
能团酸酐和游离酸酸酐。
C-O-CH 主要单官能团酸酐的性质见表5—16。
+CH:-CH
R C-O-CH2-CH-
C-O-CH1-CH O-CH:-CH
表5—16 典型的单官能团酸酐的性质
0-
OH OH
名 称 缩写
化学结构 状态
黏度(25℃)/Pa·s 熔点/℃
0
邻苯二甲酸酐 PA
由以上反应机理可以看出,酸酐和环氧树脂的固化速度受到环氧树脂中羟基浓度的影响。粉末
128
羟基浓度低则反应速率慢,羟基浓度高则反应速率快。从反应理论上来看是由一个环氧基对一个酸酐反应,而实际上仅用理论量88%~90%的酸
四氢邻苯二甲酸酐
THPA 固体
102
酐就足够了。 ②促进剂存在时 在有路易斯碱(叔胺)作为促进剂时反应机理如下所述。
a.叔胺进攻酸酐生成羧酸盐阴离子:六氢邻苯二甲酸酐
HHPA 固体
34
甲基四氢
邻苯二甲酸酐 MeTHPA
液体 0.03~0.06
H,C
b.羧酸盐阴离子和环氧基反应生成氧阴离子:甲基六氧
邻苯二甲酸酐 MeHHPA
H3C 液体
0.05~0.08
① C-NR
C-O-CH2-CH 甲基纳迪克酸酐
MNA 液体
H,C 0.138
O
c.氧阴离子与另一个酸酐进行反应再生成羧酸盐阴离子:
Cu2His-CH
十二烯基琥珀酸酐
DDSA O
液体 0.5
CH:-C O
C-O-CH2-
HO
-0
氯茵酸酐 HET
粉末 235~239
0
0 在促进剂存在时环氧树脂的固化速度不受体系内羟基浓度的支配,因此促进剂存在对低分
CH-C 子液态环氧树脂非常有效,在120~150℃能完成固化反应。在羟基含量较高的固态环氧树脂中,
顺丁烯二酸酐 MA
固体 CH-C
添加促进剂要充分注意使用寿命明显缩短的问题。促进剂存在时,酸酐作为环氧树脂固化剂的用量为实际计算值。
O
酸酐当量 酸酐用量(质量份)=
环氧当量
x100xK (1)邻苯二甲酸酐 邻苯二甲酸酐(PA)分子量148,熔点128℃,它是环氧树脂早期使用
酸酐分子量x100 的固化剂,它的特点是固化时放热量小,使用寿命长,固化产物的电性能优良,除了耐强碱性
酐官能团数x环氧当量
XK 差外,耐化学品性能良好。
酸酐分子量 X环氧值xK
邻苯二甲酸酐适宜在150℃下固化,制造大型浇注件、层压材料,邻苯二甲酸酐和环氧树脂酐官能团数
混溶可采用以下方法。先将树脂加热至120~140℃,再加人邻苯二甲酸酐,搅拌下使之完全熔
078
079
聚合物基复合材料
第5章 环氧树脂 ,对于来制,为可放和脂的液合转在1c0它时有145的适用期,标准的国化的
用邻苯二甲酸酐固化后的固化物力学、电气性能见表5—17。CH-CH-CH-CH=CH2+
150℃/6h.
表5—17 邻苯二甲酸酐固化物的力学、电气性能1.3-戊二烯
3-Me-THPA
树脂A 树脂B
CH,
项
日 35~49
80.5~87.5 CH2-C-CH-CH
拉伸强度/MPa 147~154
105~112
压缩强度/MP
105~112 126~133
异戊(间)二烯 4-Me-THPA
弯白强度/MPa 0.46
0.70 由于分子结构中有甲基存在,与羧酸碳原子较近的有一定的空间位阻,同时甲基又是供电
力学性质
冲击强度 100
100 子基团,因而有诱导效应存在,这两种作用使羧基碳原子亲电性下降,降低了酐基的活性,从
硬度洛氏M 109
一
热变形温度/℃
而使甲基四氢邻苯二甲酸酐和环氧树脂的混合物比由顺丁烯二酸酐、四氢邻苯二甲酸酐组成的3360
3150 拉伸模量/MPa
10 60
环氧树脂混合物在室温下有更长的适用期。在同样的温度(146℃)时有更长的凝胶时间,具体60
10 情况见表5-19。
顾率 0.007
0.002 0.0012
0.026 功率因数
3.64 3.65
3.89 3.50
表5—19 四种酸酐熔点及挥发性比较介电常数
16.34x10 (15.75~16.14)x10
电性质 介电强度/(V/mm)
邻苯二甲酸酐 甲基四氢邻苯二甲酸酐
四氢邻苯二甲酸酐 表面电阻系数/Ω
5.7x10■ >5.7x101
项目 顺丁烯二甲酸
体积电阻率/0·cm>8x10■
>8x10 熔点/℃
53 128
-15 102
挥发性/% 65
7 6
5 (2)顺丁烯二酸酐 顺丁烯二酸酐(MA)为白色晶体,相对密度1.509;熔点53℃;沸点
202℃。标准用量为30~40(质量份),固化条件为160℃/4h或200℃/2h。甲基四氢邻苯二甲酸酐的挥发性小,毒性也低,又是低黏度液体,和环氧树脂在室温下就
顺丁烯二酸酐熔点低,和环氧树脂配制混合物时只要预先将树脂加热到60℃,然后在搅拌能混溶,力学性能和电气绝缘强度优良,因此,它是目前用于电气绝缘的大型浇铸料、层压、
下逐渐将MA加人即可熔融,混合物在25℃下能有2~3天的适用期。缠绕环氧制品的主要固化剂。
MA的缺点是升华比PA还要严重,对操作者的眼睛、呼吸道损害较大;另外固化产物的■(5)甲基六氢邻苯二甲酸酐 将甲基四氢邻苯二甲酸酐在高压下氢化可得到甲基六氢邻苯
性很大,所以目前很少单独使用它作为环氧树脂固化剂。但MA作为混合酸酐和通过Diels二甲酸酐(MeHHPA)。其分子式为:
Alder反应制备各种酸酐的主要原料,还值得重视。
(3)四氢邻苯二甲酸酐 四氢邻苯二甲酸酐(THPA)由丁二烯和顺丁烯二酸酐按 DielsAlder反应制得的。分子量152,熔点102~103℃,不易升华,但有使环氧树脂固化物着色的倾向,在常温下蒸气压很低,因而对人体的刺激性很小。
其典型的物化性能见表5—20。
表5—18是液态酸酐和E—44双酚A型环氧树脂的固化产物性能。
表5-20
甲基六氢邻苯二甲酸酐的物化性能
表5—18 液态四氢邻苯二甲酸酐的树脂固化产物性能指
标 典型值
指
标 典型值
项
目 指
标 外观
无色至淡黄色液体 黏度(25℃)/Pa·
0.065 项
目 指
标 酯化值
675~695 蒸汽压/kPa
0.7(127℃) E—44环氧树脂100
剪切强度/MPa 1.3(144℃)
配方(质量份) 液态四氢邻苯二甲酸酐55
16~18.5 碘值/(cgl/g)
<2 /kPa
苄基二甲胺0.5 马丁耐热/℃
96~100 相对密度d■
1.168 着火点/℃
160 固化条件
100℃/1h+150℃/2h+180℃/2h 体积电阻率/Ω·cm
凝固点/℃ <-15
拉伸强度/MPa 表面电阻/Ω
9.5x101 67.5
弯曲强度/MPa 2.45x10■7
压缩强度/MPa 104
介质损耗角正切(10*Hz)甲基六氢邻苯二甲酸酐除了具有液态酸酐的黏度低、易与环氧树脂混溶、适用期长、固化
133 2.9x10-2
介电常数 放热量小、电绝缘性能好的共性之外,其最大的优点是固化产物色泽浅、耐候性好,在紫外线
2.9 (4)甲基四氢邻苯二甲酸酐
照射和长期受热状态下色泽变化很小,这与它是高纯度的脂环型结构有关,尤其是它和脂环族
甲基四氢邻苯二甲酸酐由1,3—戊二烯和异戊(间)二烯分别环氧树脂配合这种耐候性更为突出。因此甲基六氢邻苯二甲酸酐绝大多数应用于大型户外电气
和顺丁烯二酸酐进行Diels—Alder反应而制得的混合物。其最大特点是MeTHPA/环氧树脂配合
绝缘制品的浇注件和发光二极管的制造。
物的黏度非常低,而且难以从环氧树脂中析出结晶,是酸酐类固化剂使用最广泛的一种固化剂。
(6)纳迪克酸酐及甲基纳迪克酸酐 用环戊二烯及甲基环戊二烯分别与顺丁烯二酸酐反应
其反应式为:
可以制得纳迪克酸酐(NA)及甲基纳迪克酸酐(MeNA)。
080 081
聚合物基复合材料
第5章 环氧树脂
表5—21 环氧树脂的应用领域
纳迪克酸酐(nadic anhydride),化学名称为内亚甲基邻苯二甲酸酐。
分
类 应用领域
金属底漆:汽车车身、船舶、桥梁、管道等
粉末涂料:家用电器、钢制家具、管道,微型电动铁芯等涂料
无溶剂漆:线圈、变压器、特种地坪、电阻、树脂混凝土CH
罐头涂料:食品罐头、铁桶内壁等
电器:干式变压器、电力互感器,绝缘子等
浇注料
电子,电容器、变压器、印刷电路元件密封等甲基纳迪克酸酐(methylnadic anhydride),化学名称为3—甲基内亚甲基邻苯二甲酸酐,
工具:钣金模具、橡胶成型模、光侧弹性模型甲纳迪克酸酐和甲基纳迪克酸酐的结构十分相似,但前者是固体,熔点为163℃;而后者
交通工具:飞机尾翼、门,快艇、汽车车身等熔点仅为12℃,在25℃下是黏度为0.2~0.3Pa·s的液体,因此甲基纳迪克酸酐应用方便,
电子电器:发电机嵌衬、高压开关棒到青睐,它们的环氧树脂固化产物热稳定性优于邻苯二甲酸酐及甲基四氢邻苯二甲酸酐。
纤维增强塑料 设备:容器、储槽、内衬等
体育用品:球拍、滑雪板、球棒等5.3.3.4 酸酐的液体混合物及其用量计算
绝缘材料:印刷电路板、电机绝缘材料如前所述,大多数酸酐为固态,在室温下与树脂混合困难,需要在熔点以上的温度下操作,
交通工具:飞机胶黏点焊、船舶螺旋桨,汽车钣金补强等由此就产生了如下两个缺点:①在高温下酸酐会升华,产生对人体有害的刺激性蒸气;②温度
光学仪器:发电机嵌衬、高压开关棒高,会使树脂混合物的适用期缩短,这样不利于进行操作。
电气电子:元件组装、扬声器膜固定、电视安全玻璃固定等为了防止这种弊病,可将两种酸酐以一定比例混合,制成在室温为液态的混合酸酐。
胶黏剂 上木建设:混凝土预测件固定、新旧水泥连接、道路修补;路面反光器、瓷砖;旧建筑物加固、文
使用共熔混合酸酐固化环氧树脂时,每种酸酐用量的计算方法见下面的实例。物修复、设备修复
机械工业:机床维修、刀具粘接等【例】已知双酚A型环氧树脂的环氧当量190,用六氯内次甲基四氢苯二甲酸酐与六氢苯二
模压料 集成电路封装、电动机铁芯绝缘等
甲酸酐的混合物(质量比60/40)作固化剂,添加0.1%的促进剂。求100g环氧树脂的每种酸
绝缘珠、互感器、开关箱等
注射料 酐的用量。
其他 PVC的稳定剂、其他塑料的改性剂、织物整理剂等
解:
①将环氧当量换算成环氧值。泡沫材料
的夹心料、海底石油管道绝缘材料等电子工业灌封料、绝缘件、飞机导航用助燃、介电元件,飞机检验装置的夹层材料,受热结构件
环氧值=100/环氧当量=100/190=0.526②按混合比求出混合酐中每种酸酐的当量。
60g六氯内次甲基四氢苯二甲酸酐的当量=60/酸酐分子量=60/370=0.1622
40g六氯苯二甲酸酐的当量=40/酸酐分子量=40/154=0.2597③求出100g混合酸酐的当量=0.1622+0.2597=0.4219。
④求出100g环氧树脂所用酸酐量。
六氯内次甲基四氢苯二甲酸酐用量=酸酐在混合酐中的百分比x环氧值/混合酐当量
=60x0.526/0.4219=75(g)
同样,六氢苯二甲酸酐用量=40x0.526/0.4219=50(g).所以,混合酸酐用量=75+50=125(g)。
5.4 环氧树脂的应用
由于环氧树脂具有优良的特性,因此在国民经济的各个领域中被广泛地应用。无论是高新技术领域还是通用技术领域,无论是国防军事工业还是民用工业,乃至人们的日常生活中都可以看到它的踪迹。按其应用方式可分为涂覆材料、增强材料、浇注材料、模塑料、胶黏剂、改性剂.其应用领域见表5—21所列。
082
083
第6章 酚醛树脂
第6章 酚醛树脂 (5)间苯二酚 间苯二酚是无色或白色针状结晶,与甲醛反应活性高,用间苯二酚制造的
树脂可室温固化,可用于生产船龙骨和横梁,树脂的粘接力强,可用作粘接剂。6.1.1.2 醛类化合物
(1)甲醛 甲醛室温下为无色气体,—19℃液化,—118℃凝固(结晶)。温度在室温以下
时易聚合,高于100℃不聚合,气体在400℃以上分解,用于制备酚醛树脂的各种甲醛原料有气6.1 合成
体甲醛、36%甲醛水溶液(福尔马林36%)、50%甲醛水溶液(福尔马林50%)。(2)多聚甲醛 多聚甲醛为聚氧亚甲基二醇,是不同细度的白色粉末,在空气中会慢慢解
聚,受热解聚速度大大加快。多聚甲醛一般不用于树脂的生产(因价格高),但用在特殊场合,
r
如生产高固体含量树脂或低水含量树脂。多聚甲醛还可用作交联剂如作Novolak树脂、间苯二
拓展阅读 酚树脂的交联剂。
(3)三聚甲醛(三氧六环)三聚甲醛为白色晶体,有氯仿气味。三氧六环对热非常稳定,6.1.1 原料
但少量强酸能引起三聚甲醛解聚,生成甲醛。多聚甲醛或甲醛溶液(60%~65%)在2%硫酸作生产酚醛树脂的原料主要是酚类(如苯酚、二甲酚、间苯二酚、多元酚等)、醛类(如
用下加热可制得三聚甲醛。它可用作酚醛树脂的固化剂。醛、乙醛、棟醛等)和催化剂(如盐酸、硫酸、对甲苯磺酸等酸性物质及氢氧化钠、氢氧化钾
(4)乙醛 乙醛一般为40%水溶液,无色液体,有窒息性气味,能与水、醇、乙醚、氯仿
氢氧化钡、氨水、氧化镁、醋酸锌等碱性物质)。由于采用不同原料和不同催化剂制备出的酸量等混合,易燃易挥发,易氧化成乙酸,在室温下放置一段时间,会产生聚合现象,使液体发生
树脂的结构和性能并不完全相同,因此原料的选择应根据产品的性能而定。浑浊、沉淀而变质。
6.1.1.1 酚类化合物(5)三聚乙醛 三聚乙醛为无色透明液体,有强烈芳香气味,与稀盐酸共同加热或加入几
滴硫酸即分解成乙醛。(1)苯酚 纯苯酚为无色针状晶体,具有特殊的气味,在空气中受光的作用逐渐变为浅红
(6)糠醛 糠醛为无色具有特殊气味的液体,在空气中逐渐变为深褐色。糠醛除含醛基外,色,有少量氨、铜、铁存在时则会加速变色过程,因此苯酚与含铁含铜容器或反应器接触,往
尚有双键存在,故反应能力很强。苯酚与糠醛缩合的树脂具有较高的耐热性。往变色。苯酚易于潮解,苯酚含有水分时,则其熔点急剧下降,一般每增加0.1%的水,熔点将
6.1.1.3 催化剂降低0.4℃左右。
(1)合成催化剂 苯酚极易溶解于极性有机溶剂。能溶于乙醇、乙醚、氯仿、丙三醇、冰醋酸、脂肪油、松
①酸类催化剂 可用于制造苯酚甲醛热塑性树脂的酸类催化剂主要有盐酸、草酸、乙酸、节油、甲醛水溶液及碱的水溶液,但不溶于脂肪烃溶剂。苯酚与卤代烷烃作用生成醚,与酰氨
甲酸、磷酸、硫酸、对甲苯磺酸等,各种酸的性能不同,使用条件也不同,各有其优缺点。如或酸酐作用生成酯,在涂料工业中利用这个反应制得改性酚醛清漆。
使用盐酸时需要进行稀释,它的优点是价格低,在树脂脱水干燥过程中盐酸可以蒸发出去,缺苯酚的羟基为给电子基团,与苯环大π键产生共轭作用,使苯环上羟基的邻位和对位得到
点是对设备有腐蚀。 活化,即有3个电取代反应活性点。酚醛树脂就是利用这一原理合成的。
②碱类催化剂 可用于制造酚醛树脂的碱类主要有氢氧化钠、氨水、氢氧化钡,其中氢氧(2)工业酚 工业酚是从煤焦油中精馏得到,为苯酚和甲酚的混合物,其中苯酚70%、甲
化钠对酚醛的加成反应有强的催化效应,并使初级缩聚物在反应介质中有较好的溶解性,适合酚30%(甲酚有邻位、对位和间位异构体),工业酚为红棕色油状物,有毒性,腐蚀性强,稍溶
制备水溶性酚醛树脂及无水酚醛树脂。氨水可以作为苯酚苯胺甲醛树脂的催化剂,其催化性能于水,能溶于醇和醚。
较缓和,生产过程容易控制,不易发生交联,且催化剂容易除去。氢氧化钡也是一种温和的催
(3)甲酚 甲酚外观为无色或棕褐色的透明液体,工业用甲酚是在185~205℃时蒸馏煤焦
化剂,由它制得的树脂黏度低、固化速度快,且适合于低压成型。
油所得的混合物,有邻甲酚、间甲酚和对甲酚,其比例为(35~40):40:25。混合酚中的3个
除此之外,氧化镁、碳酸钠、乙酸锌、碳酸钠也可作为催化剂用于制备酚醛树脂。
组分的沸点不同,邻位易蒸馏分离,但对、间位不能蒸馏分离出来,因其沸点接近,不易分离。
(2)固化催化剂 常用固化催化剂有苯甲酰氯、对苯甲酰氯、硫酸乙酯和石油磺酸等。
生产酚醛树脂时也采用这种混合物,用邻甲酚和对甲酚与甲醛作用只能生成线型树脂,间甲酚
6.1.1.4 固化剂
有3个反应点,可以与甲醛缩聚生成热固性树脂。所以作为制造热固性酚醛树脂的混甲酚,其
(1)苯胺 苯胺又名阿尼林,为无色油状液体,极毒,暴露在空气和日光下迅速变为棕色,
间甲酚的含量应高(大于40%),间位含量越高,反应越快,凝胶时间越短,反应也越完全,缩
苯胺甲醛树脂具有良好的高频绝缘性和耐水性。
聚程度高,游离酚含量少。
(2)六亚甲基四胺 又名乌洛托品,在空气中加热可升华,可用作酚醛树脂的交联剂或固
(4)二甲酚 二甲酚为无色或棕褐色的透明液体,主要用于制造油溶性树脂,用量较少,
化剂。
有6种异构体。随结构不同,其反应活性也不一样,形成的聚合物的结构也不一样,其中
(3)三聚氰胺 又名三聚氰酰胺、蜜胺,为白色柱状结晶,用于合成树脂。
3.5—二甲酚有3个反应点,能与醛反应生成交联型树脂;2.3—二甲酚、2,5—二甲酚、3,4—二甲酚
6.1.2
加成反应 有两个反应点,与甲醛反应只能生成线型热塑性树脂;2.4二甲酚、2.6—二甲酚仅有一个反应
点,与甲醛反应不能形成树脂。
苯酚与过量甲醛在碱或酸性介质中(一般为碱性)进行缩聚,生成可熔性的热固性酚醛树084
085
聚合物基复合材料
第6章 酚醛树脂 脂,若在碱性介质(pH值=8~11)中反应,则苯酚和甲醛的摩尔比一般为617,常用催化剂
醛和苯酚的摩尔比为0.8时,所得酚醛树脂大分子链中酚环大约有5个,数均分子量在500左为氢氧化钠、氨水、氢氧化钡等,用氢氧化钠做催化剂时,反应分两步进行,即酚与醛的加成
右。若甲醛用量提高,可缩聚成分子中含有15~20个酚环的热塑性树脂。反应和羟甲基酚的缩聚反应。
分子链可进一步增长,并通过酚环对位连接起来。热塑性酚醛树脂的分子结构与合成方法有关。
生成的二酚基甲烷与甲醛的反应速率大致与苯酚和甲醛的反应速率相同,因此缩聚产物的
(1)酚与醛的加成反应 反应开始,酚与醛发生加成反应,生成多羟甲基酚以及一元酚醇和多元酚醇的化合物,这些羟甲基酚在室温下是稳定的,羟甲基酚可进一步与甲醛发生加成
一般认为在强酸性条件下对位比较活泼,缩聚反应主要通过酚羟基的对位反应,因此在热塑性树脂的分子中主要以酚环对位连接的,理想化的线型酚醛树脂应有下列结构:
反应:
HO OH
HO
CH:OH-
CH:OH
OH OH
HO
HO +CH2O→
HO CH:O
CH:OH (2)羟甲基酚的缩合反应
羟甲基酚可进一步发生以下两种可能的缩聚反应:
但也存在少量邻位结构如:OH
OH
CH2OH
HO CH:OH
-H:0 HO
OH
HO CH2
CH: CH.OH
OH
邻位结构含量随酸性增强而减少。若用高碳醛如乙醛,邻位结构也很少,应该指出的是,若甲CH2OH
醛和苯酚的摩尔比大于1时,在酸性介质条件下,反应就难以控制,最终会得到网状结构的固-H:O
OH
体树脂。 CH:OH
酸催化树脂分子量接近5000,含50%~75%的2,4'—位连接产物,其反应速率成正比于催化6.1.3 缩聚反应
剂、甲醛、苯酚浓度,与水浓度成反比。在通常加成条件下,如在较高pH值(约9)、温度低于60℃时,缩聚反应很少发生,加成
6.1.4 反应机理反应大约是缩聚反应的5倍,且甲醛与羟甲基苯酚的反应要比甲醛与酚反应容易,此现象将持
(1)强碱催化下的反应机理 在强碱性催化剂(NaOH)存在下,甲醛在水溶液中存在下续到50%甲醛被反应掉。在温度大于60℃时,缩聚反应通常发生在羟甲基苯酚、二羟甲基苯
列平衡反应: 酚、三羟甲基苯酚、游离酚和甲醛之间,反应比较复杂,在加成反应发生的同时,也发生缩聚
CH-O'+H2O→HOCH2OH 反应。由上述反应形成的一元酚醇、多元酚醇或二聚体等在反应过程中不断进行缩聚反应,使
苯酚与NaOH在平衡反应时形成负离子的形式:树脂分子量不断增大,若反应不加以控制,树脂就会发生凝胶。
OH CH.OH
+ HOCH:-
CH:OH
-CH2OCH2
CH2OH HO
HO
OH
+NaOH→
Na++H:O HO
-CH2O CH:OH
离子形式的酚钠和甲醛发生加成反应:CH:OH
OH -H2O
0 H
HO
HO
CH2OH HO
H 虽然上述两种反应都可发生,但在加热和碱性催化条件下,醚键不稳定,因此反应以后一种为
主。在此条件下,羟甲基主要与酚环上邻、对位的活泼氢反应形成次甲基(CH2)桥,而不是两个羟甲基之间的脱水反应。羟甲基苯酚之间的反应要比羟甲基苯酚与苯酚的反应快。
在酸性反应条件下,苯酚和甲醛在溶液中加成形成羟甲基苯酚后,再与苯酚进行缩聚反应,缩聚反应速率比加成反应速率快,约5倍以上。Knopf和Wagner用NMR证实羟甲基苯酚在酸
上述反应的推动力主要在于酚负离子的亲核性质。性溶液中以羟甲基酚阳离子的形式存在。实验还表明,羟甲基苯酚与Novolak链端基团的反应
对羟甲酚可通过下列历程形成:活性要比链内基团高。正因为这样,在酸性缩聚反应中,支化反应是相当少的。当苯酚和甲醛
HO 摩尔比在0.85~0.87以上时,随着聚合反应的进行,聚合物的浓度增大,单体浓度减少,情况
发生变化,内取代反应发生,从而导致发生凝胶。在这种凝胶物中,可萃取物含量是相当高的,很显然交联程度不高。要使交联产物有优良的性能,需加大量交联剂,如六亚甲基四胺,当甲
086
聚合物基复合材料
第6章 酚醛树脂 邻对位比取决于阳离子和pH.对位取代用K+,Na+和较高的pH时有利,而邻位取代在
表6-1
三大热固性树脂的特点低pH、用二价阳离子如Ba+、Ca2+和Mg2+时有利,邻位的酮式结构因位阻及氢键而较对位
酚醛树脂
环氧树脂 难于形成。其反应动力学还未完全弄清楚,一般认为是二级反应即取决于酚盐浓度和甲二醇液
容易制成B阶树脂,有优良
固化收缩小,随固化剂种类而异,
不饱和树脂 度,反应速率=k[pH][甲二醇](但对氨催化反应与此不同,是一级反应)。Freeman 和 Lewis
的预浸渍制品的特性
体积收缩1%~50%
100%固化
固化时无挥发性副产物,几乎已达到些反应如图6—1所示。其中有些反应结构还不完全清楚,如甲二醇是如何与酚氧离子反应的。
研究了30℃下酚醛的反应,其配比P:F:NaOH=1:3:1(摩尔比)。假定其为二级反应,
固化物耐高温特性,特别是高
固化物机械强度高 温强度比聚酯好得多
尺寸稳定性好
可用多种手段实现固化,如过氧化物。固化迅速,即使在常温下也能固化
有优良的耐燃性
黏结性好 甲醇化苯酚与甲醛反应速率要比苯酚与甲醛快(2~4倍),因此尽管甲醛与苯酚之比高达3:1,
固化物强度比聚酯高
电性能、耐腐蚀性能(特别是耐碱
可低压成型,接触压成型也可紫外线,射线等
苯酚的残留率仍然较高(Resool树脂中)。
可用水和醇的混合溶剂,操作
若对树脂及固化剂进行选择,能得
力学性能及电性能优良热变形温度高,脱模时变形小
性)优良 HO
HO
耐药品性好 优点
方便
到耐热性好的固化物
能赋予柔软性、硬质、耐候性、耐热性、CH.OH
HOCH
CH.OH
成型只需加热、加压、不需添
固化物无臭味,能用于食品行业
耐药品性、触变性、难燃、耐熄等特性加引发剂和促进剂
树脂保存期长,选择固化剂可以制
可着色,获得透明美现的涂膜价格低廉
成B阶树脂,有良好的制预浸渍制
能实现兼具保护与装饰的涂装品的特性
固化时放热,使温度上升(这一点可在HOCH
CH.OH
冷模压中获得应用) 固化时不会像聚酯那样,容易受空
气中的氧的阻聚
能实现空气干燥 CH.OH
CH2OH
持稳定,缠绕特性好
不含挥发性单体,配合组成时常保
固化收缩非常小,甚至能达到零收缩固化比聚酯慢,到完全固化需
固化剂毒性太大,操作应十分注意
一般来说,空气中氧的存在会妨碍固化CH.OH
CH2OH
较长时间
固化时间比聚酯长,达完全固化必
硫黄、酚类化合物、炭等混入时,固化困难图6—1 甲酚醇化的反应
黏度高,浸渍玻璃纤维需一定的
影响 固化时有副产物产生,成型时
须进行长时间的热处理
特殊的金属或化合物对固化有很大的需比聚酯更高的温度和压力
(2)强酸催化下的反应历程 通常认为,酸催化下的反应是与甲醛或它在水溶液中的甲二
一般讲,固化物硬而脆,但经
时间
通常有百分之几的固化收缩醇形成的质子性质有关的亲电取代反应。
过改性有可能做到半硬质状态
固化放热峰高
固化方法不当时,由于固化放热及收缩缺点
固化物的颜色在褐色与黑色
价格较高
不理想,在制品中会产生裂纹前一步反应比较慢,是反应速率的决定步骤,后一步反应比较快,邻位反应也可发生,但
之间,不能随意着色或着淡色
固化易受温度,湿度的影响间位反应不发生。研究证明羟甲苯酚在酸性条件下是瞬时中间产物(但确实存在),很快脱水。
耐腐蚀性好,但耐候性差,日
制造后随时间变化固化特性等也容易脱水的碳锚离子立即与游离酚反应,生成H+和二酚基甲烷。
久会变色
产生变化 动力学数据表明,H+在酚和醛反应的开始阶段是活泼的催化剂,缩聚反应速率大体上正比
低温储存
黏稠性液体有特殊的臭味于氢质子的浓度。
预浸渍制品的保存期短,必须
易燃 若甲醛和苯酚的摩尔比等于1,则可导致支化,甚至出现凝胶,这时测得的临界支化系数为
常用热固性树脂的性能见表6—2。0.56,即在反应程度达56%时就会出现凝胶。反应动力学研究表明,反应级数为二级(多数情
况),反应速率与[H+]成正比,整个反应活化能和活化熵随pH提高而增加,表明机理发生变化。
表6—2 常用热固性树脂的性能热塑性酚醛树脂即二阶树脂是可溶、可熔的,需要加入诸如多聚甲醛、六亚甲基四胺等固
性 能
酚醛树脂
不饱和树脂
环氧树脂
有机硅树脂 化剂才能与树脂分子中酚环上的活性点反应,使树脂固化。热固性酚醛树脂也可用来使二阶树
密度/(g/c㎡)
1.30~1.32
21~49 脂固化,因为它们分子中的羟甲基可与热塑性酚醛树脂酚环上的活泼氢作用,交联成三维网状
拉伸强度/MPa
42~64
5.0 1.10~1.46
1.11~1.23
1.7~1.9 结构的产物。
断裂伸长率/%
1.5~2.0
5.0 42~71
85 6.2 性能
压缩强度/MPa
88~110
92~190 拉伸模量/GPa
3.2
2.1~4.5
3.2 11
64~130 弯曲强度/MPa
78~82
60~120
130
69 酚醛树脂有以下主要特征:①原料价格便宜、生产工艺简单而成熟,制造及加工设备投资
线膨胀系数/(x10—6/℃)热变形温度/℃
78~120
308 60~120
120 少,成型加工容易;②抗冲击强度小,树脂既可混入无机或有机填料做成模塑料来提高强度,
09 60~80
80~120 洛氏硬度
120
115
4~8 100
45 也可浸渍织物制层压制品,还可以发泡;③制品尺寸稳定;④耐热、阻燃,可自灭,燃烧时发
体积电阻率/Ω·cm
1012~101
101~107 收缩率/%
8~10
4~6
1011~10■ 1~2
烟量较小且燃烧发烟中不含有毒物质,电绝缘性能好,在电弧作用下会生成炭,故耐电弧性不
介电强度/(kV/mm)
10 佳:⑤化学稳定性好,耐酸性强,由于含苯酚型羟基,因此不耐碱;⑥长时间置于高温空气中
介电常数(60Hz)
6.5~7.5
3.0~4.4 15~20
16~20
7.3 14~16
4.0~5.0 会变成红褐色,故着色剂使用受到限制。酚醛树脂和其他树脂的一些特点列于表6—1.
介电损耗角正切(60Hz)
0.10~0.15
0.003
3.8 0.001
0.006 088
089
聚合物基复合材料
第6章 酚醛树脂 续表
大多数聚合物材料都是可燃烧的,但可以通过添加阻燃剂来改变,可达到V—1和V—0级。不饱和树脂
环氧树脂
有机硅树脂
酚醛树脂是例外,它既具有阻燃性,又具有低烟释放和低毒性。酚醛树脂主要由碳、氧和氢组酚醛树脂
課 性
125
50~80
成,它们的燃烧产物与燃烧条件有关,主要是水蒸气、二氧化碳、焦炭和一氧化碳(中等量).100~125
0.15~0.6
0.14
低
因此燃烧产物的毒性相对较低。毒性与酚醛树脂的分子结构有关,研究表明改性酚醛树脂的复耐电弧性/
■水率(24h)/%
0.12~0.36
良好
优良
差
合材料具有最低的毒性,酚醛燃烧时易形成高碳泡沫结构,成为优良的热绝缘体,从而制止内对玻璃、陶瓷、金属的黏结性
优良 耐化学性
轻费
无
轻微
部的继续燃烧。交联密度高的树脂,有利于减少燃烧时毒性产物的放出,因为低分子量酚醛分经微
侵蚀
侵蚀
子易分解和挥发。酚醛树脂的发烟特性与氧指数还与残碳率有关,氧指数高则残碳率高,它们国籍
侵蚀
侵蚀
轻微
之间存在线性关系。残碳率也与酚醛树脂的酚取代有关,非取代酚的酚醛树脂的残碳率往往高强酸
轻微
无 轻微
非常轻微
降解 第
降解
降解
于取代酚的酚醛树脂,见表6—4,酚醛树脂还可使用阻燃添加剂来提高树脂的阻燃性,中等燃烧强碱
侵蚀
侵蚀
某些有机溶剂
能力的填料或增强纤维,如纤维素、木粉等可作为阻燃添加剂。较理想的阻燃添加剂有四溴双有机溶剂
有些有机溶剂
酚A(TBBA)和其他溴化苯酚、对溴代苯甲醛,无机和有机磷化合物如三(2—氯乙基)磷酸酯、6.2.1 基本性能
磷酸铵、二苯甲酚磷酸酯、红磷、三聚氰胺及其树脂、脲、二氧二胺、硼酸及硼酸盐等及其他酚醛树脂与其他热固性树脂比较,其固化温度较高,固化树脂的力学性能、耐化学腐蚀性
无机材料。 可与不饱和聚醋相当,但不及环氧树脂;酚醛树脂的脆性比较大、收缩率高、不耐碱、易潮、
表6—4 各种酚醛树脂的氧指数和残碳率电性能差,不及聚酯和环氧树脂。
氧指数
残碳率/% 酚醛树脂与不饱和聚酯、环氧树脂相比,酚醛树脂的马丁耐热温度、玻璃化转变温度均比
酚
Novolak
Resol
Resol Novolak
前两者高,尤其是在高温下,力学强度明显高于前两者,在300℃以上开始分解,逐渐炭化,
34~35
36
56~57
54 苯酚
800~2500℃在材料表面形成炭化层,使内部材料得到保护,因此广泛用作火箭、导弹、飞机、
间甲酚
33
51 飞船上的耐烧蚀材料。
间氯代苯酚
75
74
50
50 间溴代苯酚
75
76
41 6.2.2 热性能及烧蚀性能
91 酚醛树脂的耐热性是非常好的。酚醛树脂的玻璃化转变温度、马丁耐热等均比不饱和聚酯
6.2.4 耐辐射性和环氧树脂高,酚醛树脂及其玻璃纤维增强材料的模量在300℃内变化不大,虽然弯曲强度在室
无填充的酚醛树脂耐辐射性相对较低,而玻璃或石棉增强的酚醛树脂是非常好的耐辐射合温下不及聚酯和环氧树脂,但在高于150℃下,强度都比它们高。
酚醛树脂在300℃以上开始分解,逐渐炭化,而成为残留物,酚醛树脂的残留率较高,可在
成材料,但酚醛树脂的氧含量对耐辐射具有相当不利的影响。当高能辐射(y射线,X射线、中60%以上。酚醛树脂在高温800~2500℃下在材料表面形成炭化层,使内部材料得到保护。因此
子、电子、质子和氘核)通过物质时,在原子核内或在轨道电子内出现强烈的相互作用使大部酚醛树脂广泛用作烧蚀材料,用于火箭、导弹、飞机、宇宙飞船等。
分入射能损耗。这种作用的最后结果是在聚合物材料内形成离子和自由基,从而破坏化学键,6.2.3 阻燃性能和发烟性能
数决定着耐高能辐射性;含有芳环的聚合物具有低得多的降解速度(因为瞬时活性种的共振稳并同时伴随着新键的形成,紧接着以不同的速度发生交联或降解。破坏和形成键的相应速度常
酚醛材料燃烧时形成高碳泡沫结构,成为优良的绝热体,从而阻止了材料内部的燃烧。酚
定);通常刚性高分子结构耐热固性材料要比柔性热塑性和弹性体结构更耐辐射。阻碍效应主要醛材料的燃烧产物主要是水、二氧化碳、焦炭和中等含量的一氧化碳,燃烧产物的毒性较低。
是离子或自由基的结合,加少量某些物质具有很好的稳定效应,类似抗氧剂,但稳定机理仍需酚醛树脂复合材料具有不燃性、低发烟率、少或无毒气体放出,在火中性能如可燃性、热释放、
研究。耐辐射性可通过加矿物填料来改善;相反,加一些添加剂(称电波敏感剂)可加速损坏,发烟、毒性和阻燃性等远优于环氧树脂和聚酯树脂、乙烯基酯树脂。表6—3列出几种树脂的发
如在酚醛树脂中加入纤维素可加速材料的辐射破坏。烟情况,可见酚醛树脂的发烟密度明显较低。不仅如此,酚醛材料还具有优良的耐热性,在
由于酚醛树脂尤其是复合材料具有优良的耐辐射性,且具有高的耐热性,故酚醛模压塑料300℃下1~2h仍有70%强度保留率。
用作核电设备和高压加速器的电学元件、处理辐射材料的装备元件、空间飞行器的结构组件,表6—3 几种树脂燃烧时的发烟密度
以及用作核电厂的防护涂料。住
脂
发烟密度 网烟火
火
6.3 应用及发展 酚醛树脂
2 环氧树脂
132~206
16 乙烯基树脂
39
482~515
6.3.1 应用 聚氯乙烯
144
530 364
除了用作酚醛复合材料的树脂基体外,酚醛树脂大量用于胶黏剂、涂料、离子交换树脂、感光性树脂、酚醛纤维、电流变体、催化剂等。
090
091
聚合物基复合材料
第6章 酚醛树脂 耐热性胶黏剂主要分为以下几种;丁腈—酯醛胶、聚乙烯醇—酚醛胶、氯丁—酚醛胶、氟橡胶、
表6—5 酚醛模塑料的典型特性酚醛胶、酚醛—缩醛—有机硅胶、酚醛—环氧胶等。
种
特
点 除料分为水溶性和油溶性两种。涂料工业中主要使用油溶性酚醛树脂,其优点是干燥快、
涂膜光亮坚硬、耐水性及耐化学腐蚀性好;缺点是容易变黄、不宜制成浅色漆、耐候性差,主要用于防腐涂料、绝缘涂料、一般金属涂料、一般装饰性涂料等方面。水性酚醛树脂胶黏剂中
日用品(R)
用
途 综合性能好,外观,色泽好
日用品、文教用品,如瓶盖,组扣等电气类(D)
具有一定的电绝缘性
低压电器、绝缘构件,如开关、电话机壳,仅表壳绝缘类(U)
电绝缘性、介电性较高且成本低
电信、仅表和交通电气绝缘构件游离酚含量较低,在合成中不使用有机溶剂,对人体危害较小。其优点是粘接力强、耐热性好、
高频类(P)
有较高的高频绝缘性能
高频无线电绝缘零件、高压电气零件、超短波电信,无耐水、耐老化,主要用于制造耐水胶合板、建筑模板、船舶板、纤维板等。
线电绝缘零件 在线型酯醛树脂中加人芳香族重氮化合物或重氮盐化合物,可作为感光性酚醛树脂使用,
高电压类(Y)
介电强度超过16kV/mm
高电压仪器设备部件 耐酸类(S)
较高的耐酸性
接触酸性介质的化工容器、管件、阀门用于印刷平板的制作。谭晓明等研究了紫外线引发剂安息香乙醚(BE),2,2—二乙氧基苯乙酮
无氨类(A)
使用过程中无NH,放出
化工容器、纺织零件、蓄电池盖板、瓶盖等(DEAP),2—丙基硫杂惠酮(ITX),空气中的氧、胺增效剂乙基—4(甲基—氨基)苯甲酸酯
湿热类(H)
在湿热条件下保持较好的防霉性、
热带地区用仪表,低压电器部件,如仅表外壳,开关在外观和光泽
较高温度下工作的电器部件(EDAB)和硅胶对含丙烯酰基和季铵盐基酚醛树脂(PreP)感光性能的影响。结果表明,光引
马丁耐热温度超过140℃耐热类(E)
水表输承密封圈、煤气表具零件发剂ITX的引发效率最高,稳定性好;氧对PreP与交联型稀释单体的感光交联反应有阻聚作
耐冲击类(J)
用纤维状填料,冲击强度高用:EDAB能抑制氧的阻聚作用;硅胶可提高抗湿性和表面黏性;PreP可溶于水、无水乙醇和
耐磨类(M)
耐磨特性好,磨耗小 苯的混合溶剂,在180℃下受热1h有57%的季铵盐分解。张拥军等在水溶液中制备了酚醛树脂
特种(T)
根据特殊用途面定 与重氮盐(PR—DS)、重氮树脂(PR—DR)的复合物。两种复合物都有很高的光敏性,在紫外线
作用下,PR—DR的离子键转化为共价键从而不溶于DMF,可作为光成像材料。
(3)纤维增强酚醛复合材料 纤维增强酚醛复合材料具有优异的性能,可替代金属用于汽在酚醛树脂上导入—SO3H、—CH2COOH、—COOH等基团,可制得酚醛型阳离子交换
车和机器制造业,适用于水泵外壳、叶轮、恒温箱外壳、燃料输送泵、盘式制动器活塞、整流树脂,应用在工业污水处理、石油化工工艺等方面。
子、带燃料导管和回气导管、三角皮带盘、齿轮皮带、阀盖、整流器、滑轮、导向轮等,也做将热塑性酚醛树脂用甲醛的浓盐酸溶液进行热处理后,甲醛与酚醛树脂分子发生交联,再
井下用机械零件、汽车零件如 Resinoiod 公司Resinoiod 系列,其中 Resinoiod 1382含40%玻璃 进行熔融纺丝可制得酚醛纤维。尽管其摩擦性能不太好,但在加热下不会发生软化,适合做消
纤维,弯曲强度110.3MPa,拉伸强度79.3MPa,缺口冲击强度117.3J/㎡2,可压缩、注射成防服、焊接工作服、安全手套等。2003年,德国汉堡的欧洲酚醛纤维公司宣布研制成功纤度为
型。类似产品有Rogers公司RX630,Durez公司的Durez 31988、Durez 31735等。Perstorp 1.65dtex(1dtex=10—6kg/m)的酚醛纤维(Kynol)。这种经交联的酚醛纤维具有高防火性、耐
Ferguson公司的A2740是55%玻璃纤维增强的粒状酚醛塑料,弯曲强度260MPa,模量22GPa,腐蚀性、舒适性,主要用于飞机、铁路、船舶和汽车的隔热和绝缘,防火队员的座椅,防火和
拉伸强度100MPa,模量19GPa,缺口冲击强度4.5~5.5J/㎡,热变形温度230℃,热导率防化学服装的衬里,极地的防寒材料,水箱的衬里,潜水艇的垫罩,飞机和舰船的逃逸盖,各
0.35W/(m·K),潜在应用是汽车、飞机、国防和电子工业。德国Raschig公司的玻璃纤维填充种复合材料、包装物、刹车片和联轴节等。Kynol织物对安全性要求高的地方,如汽车、飞机、
酚醛模塑料Resinol PF4041具有很高的刚性,在185℃下具有高韧性和断裂延伸率,用于汽车中机场、饭店、医院、护理院、渡口、潜水艇和火车等十分有用。
水泵壳体和叶轮。日本Kobe Steel Works开发了30%碳纤维增强酚醛模塑料,可替代不锈钢板,酚醛树脂可用缠绕、RTM、注射成型、模压成型等加工,也可发泡成型,其耐热、耐磨、
其相对密度为不锈钢的1/5(为1.4),具有优良的耐磨性和自润滑性,可用于轴承等。耐化学、尺寸稳定、电性能良好。酚醛树脂的初始应用主要在电气工业,用作绝缘材料,替代
6.3.2 最新发展当时应用的传统材料如虫胶、古塔胶。由于其质轻、容易加工而获得广泛应用,可替代木头、
金属,成为20世纪前半世纪的重要合成聚合物材料,用于电吹风、电话机、壶把柄等日用品,也用在建筑、汽车等工业领域,主要应用为模塑料和层压板,用作绝缘材料等。
8 (1)酚醛模塑料 如前所述,酚醛树脂固化后机械强度高、性能稳定、坚硬耐磨、耐热、
阻燃、耐大多数化学试剂、电绝缘性能优良、尺寸稳定,且成本低,是一种理想的电绝缘材料,
拓展阅读 广泛应用于电气工业,故酚醛塑料又俗称“电木”。酚醛模塑料的典型特性见表6—5。
后来,一些应用被更容易制造的热塑性塑料替换,但酚醛树脂也找到了一些新的用途,且
6.3.3 回收利用在某些场合还没有理想的材料可替代,如烧蚀材料。目前世界酚醛树脂主要用于木材加工工业、
热固性树脂及其复合材料因具有轻质、高强、比强度高等优异性能,被广泛应用于各行各热绝缘和模压料,约占总量的75%。在美国60%用于木材工业,15%用于纤维绝缘、9%用于
业。然而,怎样有效处理和利用其废弃物也是一个问题。据统计,全世界的复合材料的年产量模压料。酚醛树脂用量与聚氨酯和聚酯相当。最近几十年,注射酚醛模塑粉进展很快,它比压
超过500万吨,其废弃物达100万吨,回收利用率仅为10%。缩模塑料更经济、生产期更短、机械化程度更高。
我国玻璃钢产业自1958年开始发展以来,至今已具有一定的生产规模。据统计,2001年我(2)酚醛塑料 酚醛塑料具有耐高温、耐冲击、低发烟和耐化学性,且成本低等特点,使
国玻璃钢/复合材料年产量约45万吨。若玻璃钢产品的使用寿命按20年计算,1980年前后的玻酚醛树脂有较快的发展,现正与热塑性塑料相竞争,如酚醛塑料在汽车燃料系统部件中正取代
璃钢制品,均将陆续进入更新的时期。而且我国80%左右的复合材料制品为手糊生产,生产中聚苯硫醚和聚酰胺(尼龙)等热塑性塑料。若用于模制嵌件,酚醛塑料部件在受力时具有优异
产生的废弃物更多,且回收利用率很低。的抗变形能力,性能也可靠。
092
热固性复合材料废弃物的回收和利用是收集热固性复合材料生产、使用过程中产生的热固
093
聚合物基复合材料
第6章
酚醛树脂 性复合材料度到物,对之采取物理管样、化学分解、生物解等方法,回收其中的各种有
的提高。粉碎回收法由于方法简单,可回收的热固性复合材料废弃物品种较多,对用一般方法分或其能使之实或指外预则上选择在山内或宽地,也有些单位采取就近掩理,这种方法造
难以回收的热固性复合材料废弃物(如SMC废弃物)也能较好地回收,且不会对环境造成污
比较简单,不会造成土地浪费,但由于燃烧中产生大量毒气,同样造成环境污染。
需,不会道成家,热固性复合材料回收利用技术日益受到关注,同收加工多以粉醉种染,所以最为常用,但其缺点是要消耗大量能量。
总之,国外许多生产厂商的研究试验及生产实践已经证明,玻璃钢复合材料包括酚醛复合材料制品是可以回收再生的。但在我国关于玻璃钢边角废料的回收利用尚为空白,还没有一个
解法在再生热剧性复合材料废弃物的处理新技术:二是开发可再生、可降解的新材料,回收方解法技术为主,已具备一定的规模,技术日趋成熟。其主要研究方向大致分为两个方面,一
回收加工边角废料的企业或场所,也没有实施边角废料回收利用的计划等。目前我国玻璃钢产业的环境污染问题虽然尚没有国外的那么严重,但也必须引起有关部门及玻璃钢业界的重视。
21世纪是环保的世纪,随着我国可持续发展战略的实施,对环境保护提出了更高的要求。法有物理方法和化学方法两种。
我国2000年1月份开始实施的《中华人民共和国固体废弃物污染环境防治法》提出,国家鼓(1)化学法
励、支持开展清洁生产,减少固体废弃物的产生量。西欧各国有关的环保当局也曾明令,如不①焚烧法 利用度弃物作燃料进行焚烧,以获取能量。能量回收技术有液体床技术、旋转
炉技术和材料燃烧技术等。热塑性玻璃钢所含能量较高,适用于这一方法。但热固性玻璃钢饲解决玻璃钢复合材料的利用问题,将限制其发展。如何解决玻璃钢复合材料废弃物处置问题已
如汽车中用量最多的SMC,其有机物含量和所含能量较低,而灰分含量很高。灰分通常采用填成为我国乃至全世界玻璃钢复合材料工业界当前面临的一个十分紧迫的课题,是21世纪对玻璃
钢复合材料工业的挑战,对玻璃钢复合材料事业的生存与发展具有重大而深远的意义。埋的方法处理。
②热解法 热解法是将一种物质在无氧的情况下利用高温(不燃烧)变成一种或多种物质的方法。用高温分解的方法来回收利用热固性复合材料制品有较大的难度,费用较高。但回收利用的效果较好。热解法适用于处理被污染的废弃物,例如处理热固性复合材料部件。
在无氧的情况下,高温分解使热固性复合材料废弃物分解成燃气、燃油和固体3种回收物。其中每一种回收物都可以进一步回收利用。该工艺设备由原料处理及喂料系统、高温分解反应器、提纯和洗涤系统、控制系统和出料系统组成。回收的燃气可用来满足热分解的需要。多余的燃气通过管道可供锅炉及内燃机使用。固体副产物可用作SMC、BMC、ZMC和热塑性塑料的填料。
热固性酚醛树脂是不溶不熔的高分子材料,可以裂解再加以利用。酚醛树脂在440~500℃进行加氢分解时,液化率为30%,其中40%~50%是苯酚,用活性炭载附白金作催化剂时,液
专
化率可达到80%以上。在722℃裂解时,产物有24.3%的气体、15.8%的有机液体、9.2%的
在部
水、42.2%的炭黑、9.5%的灰分。有机产物包括脂肪族烃5.24%、芳族化合物2.44%、苯酚8.25%和少量其他物质,气体是58.4%的二氧化碳和其他可燃气体。
名
酚醛树脂热解后可产生活性炭。在600℃裂解30min,即碳化成碳化物,用盐酸溶解掉其中日■
核日期
总日码
的灰分,增大碳化物的表面积,在850℃高温下,用水蒸气活化,可得到吸附力强的活性炭,产
(5
率12%,比表面积1900㎡2/g,对十二烷基苯磺酸钠的吸附能力比通用活性炭高3~4倍。211-12
2-
11-1-35
核载质
011-1■ Yoshikai 以酚醛树脂废料和玻璃纤维增强酚醛树脂废料为原料,用单螺杆挤出机成型制备
200-320
0
2
21-1
出燃料。酚醛树脂废料热循环利用中出现的低生热值和飞灰现象可通过与聚丙烯废料共混加以
(-1
日 2号
解决。对其组分分析、元素分析和生热分析表明,该材料表现出适合做燃料的性质。这种方法
0-051
000-055 01-
有望成为热固性材料循环利用的一种途径。
1000.0
000000 出日期
(2)物理方法 物理回收是直接利用热固性复合材料废弃物并不改变化学性质的方法,是
20-1
001-
将废弃物破碎并碾磨成细粉再进行回收。回收设备主要是由废料输送机、成粒粉碎机、鼓风机旋风分离器、定量供料箱、分级设备和集尘机等组成。粉碎后碾磨成的细粉含有一定量的玻璃纤维。它的分散性很好,可制得具有高附加值的增强型材料。
美国大豆研究会(United soybean)用大豆蛋白和酚醛树脂胶黏剂,以木纤维、麻纤维等废纤维作增强纤维和填料,制成可循环使用的酚醛复合材料,经过挤出、模塑成型后,强度高,毒性小,是良好的建筑材料。
美国Asphalt研究中心已对这种回收酚醛粉料的使用价值进行了充分的技术经济评估,认为这些粉料与目前使用的填料性能相似,而且利用这些混合料制成的制品的工程使用性能有明显
094
095
第7章
氟酸酯树脂
OCN 第7章 氰酸酯树脂
氯酸酯树脂(samnte rein,CE)通常定义为含有两个氧酸酯基(OC=N)聚苯喹啉氰酸酯
二元酚行生物,其通式为:N=CO—Ar—OC=N,商品化的氰酸酯的结构式可表示为;R
0-Ar-OCN
聚健酮氨酸酯
其中,R可以是氢原子、甲基和烯丙基等,X可以是亚异丙基、脂环骨架等。
氨酸酯树脂在热和催化剂作用下,会发生环三聚反应,生成含有三嗪环的高交联密度网络结构的大分子,固化氰酸酯树脂具有低介电常数(2.8~3.2)和极小的介电损耗(0.002~0.008)、高玻璃化转变温度(240~290℃)、低收缩率、低吸湿率(<1.5%)以及优良的力学性能和粘接性能等特点。总体面言,氰酸酯树脂具有与环氧树脂相近的加工性能,具有与双马
CH1
来酰亚胺树脂相当的耐高温性能,具有比聚酰亚胺更优异的介电性能,具有与酚醛树脂相当的
聚■枫氰酸酯
耐燃烧性能。尽管氰酸酯树脂是出现较晚的高性能树脂,但它在复合材料领域上的应用取得了成功,例如高性能印刷电路板和飞机雷达罩。目前已经开发的氰酸酯树脂主要应用于3个方面:
NCO
高速数字及高频用印刷电路板、高性能透波结构材料和航空航天复合材料用高性能树脂基体。氰酸酯加热到150~200℃,可发生三聚反应形成三嗪结构,随反应程度的不同,可以控制
线型酚醛氰酸酯 预聚物为液体、固体,也可以制成溶液。氰酸酯树脂具有与环氧树脂相似的加工工艺性,可在
177℃下周化并在固化过程中不产生挥发性小分子。氰酸酯与其他热固性树脂的性能比较见表
NCO
7—1.目前,商业用的氰酸酯除双酚E氰酸酯(二氰酸酯基二苯基乙烷)是低黏度液体外,其他氰酸酯均为结晶性固体,但是这些晶体的熔点都低于制备它们的酚类化合物的熔点,使氰酸酯树脂有较好的工艺性。图7—1为几种新型氰酸酯树脂的结构。
表7—1 热固性树脂基体的比较
氨酸酯—XU71787.02L
树脂性能 环氧树脂
酚酸树脂 增韧 BMI
氰酸酯树脂 密度/(g/cm')
1.2~1.25 1.24~1.32
1.2~1.3 1.1~1.35
使用温度/℃ 室温-180
200~250 约200
约200(250) 拉伸模量/GPa
3.1~3.8 3~5
二(4—苯氧基氰基苯基)苯基磷氧化物介电常数(1MHz)
3.8~4.5 4.3~5.4
3.4~4.1 3.1~3.4
固化温度/℃ 室温~180
90~150 3.4~3.7
2.7~3.2 固化收缩
0.0006 220~300
180~250 起始分解温度/℃
260~340 300~360
0.0007 0.004
0.002
360~400 400~420
NCO 二(4—苯氧基氰基苯基)苯砜
Ph
图7—1 新型氰酸酯树脂的结构
Ph
OCN 7.1 合成
聚苯亚苯氰酸酶
氰酸酯单体不能通过氰酸与烷烃直接反应来制备,其反应产物是异氰酸酯。人们研制了多
096 097
聚合物基复合材料
第7章 氟酸酯树脂 种选径实施氧限后公司于1963年申请的专利,与其他制备路线不同的是,该方法可以在工业
采用这种合成方法的好处在于;可以省去制备易于挥发或升华、有剧毒的卤化氰,使总体最利提力制备单酸、多酚和一系列部分古化的脂肪族羟基化合物的衍生氧酸酯,也可以用于
工艺一步化、简单化,但是这又大大增加了终产物氰酸酯的提纯难度。
备一系列芳基氰酸酯、卤烧基氰酸酯,但不能制备烷基氰酸酯。
7.1.4 硫三唑的热解反应我国学者对须酸酯树脂的应用也进行了一些研究工作,到目前为止,除西北工业大学与教
Jesen和Holm 曾通过硫代氨基甲酸酯与重金属氧化物反应,消去硫化氢制备氰酸酯,但这空工业总公司联合合成的双酚A型解酸酯树脂外,国内还未见到有关氰酸酷树脂合成的其他报
种方法的产率只有40%~57%,此外还可用硫酰氯制备硫三唑,再裂解制得氰酸酯,该方法可道,氰酸酯单体的合成方法如下。
用于制备脂肪族氰酸酯,反应式如下:7.1.1 酚类化合物与卤化氰的反应
R-OH+C1- 在碱存在的条件下,卤化氰与酚类化合物反应制备氰酸酯单体。
ArOH+XCN→ArOCN+HX
反应式中的X可以是C1、Br、I等卤素,但是通常采用在常温下是固体、稳定性好、反应7.2 性能
活性适中和毒性相对较小的溴化氰:ArOH可以是单酚、多元酚,也可以是脂肪族羟基化合物,
反应介质中的碱通常采用能接受质子酸的有机碱,如三乙胺等。反应在0~20℃下的有机溶剂中在氰酸酯分子中,与—OCN连接的碳原子具有强烈的亲电性,因此,氰酸酯在温和的条件
进行,根据各种酚的结构不同而反应温度各有差异。下可与亲核试剂反应,可以与多元醇、胺和羧酸反应,其产物与异氰酸酯的反应产物不同,可
最常用和简单的氰酸酯是双酚A氰酸酯,其制备反应可表示如下:形成酰胺碳酸酯、异脲等。氰酸酯没有异氰酸酯活泼,反应产物不及异氰酸酯稳定,无论是水
CH CH:
解稳定性,还是化学稳定性和热稳定性都如此。
■2
HO -OH+2CICN
NCO OCN+2HCI
7.2.1 反应性 CH:
CH
双酚A与溴化氰的反应温度通常控制在—5~5℃之间,而酚醛与溴化氰的反应温度要控制在芳基氰酸酯不能重排形成芳基异氰酸酯,可进行一系列的反应。由于—OCN基上O、N的
—30℃左右。该法合成氰酸酯树脂单体的过程中,主要有两类副反应发生,一是因为合成反应电负性较大,C具有很强的亲电性,甚至在较温和的条件下,氰酸酯也可以与亲核试剂反
是在碱性环境下进行的,因此有少量的氰酸酯单体在碱的催化下发生二聚反应生成非晶态的半应。—OCN上的O也可以发生亲核加成反应,氰酸酯可能的反应可分类如下。
固体状的氰酸酯低聚物,同时,在碱性条件下体系中含有的少量水分或合成原料酚本身与反应(1)亲核反应 -OCN基团中的C=N可与活泼氢如ROH.RSH.R'RNH.HON-CRR'
生成的氰酸酯继续反应生成氨基甲酸酯或亚氨基碳酸酯等,这将影响合成产物的储存稳定性和等反应。如氰酸酯与取代胺的反应:
终产品使用性能如耐热性和耐水解性等。这种方法合成氰酸酯的方法已用于工业化生产,工艺
HN
路线简单,合成产率和产品纯度高,且生产的芳香族氰酸酯的稳定性极好,由它们制造的最终
R-OCN+HNRR'→R-O-C-NH-R
产品使用性能优异。
(2)亲电加成反应 氰酸酯可与酸酐反应,生成亚氨基甲酸酯。
7.1.2 酚盐与卤化氰反应
R-OCN+O
最早合成氰酸酯的方法是用碱酚盐(如酚钠)类化合物与卤化氰反应(M为金属钠或金属钾):
MO-Ar-OM+XCN→NCO-Ar-OCN+MX (3)1,3-偶极加成反应 氰酸酯可以与NaN3,CH2-N-N、C2HsCOO-CH-N=N、
CsHs-C(CI)=N-NH-C6H5、R-CH-N(O)-R'、Ar-CNO等发生1,3-偶极加成反应。
在该合成方法中,反应生成的氰酸酯很容易在强碱性催化条件下发生三聚反应以及与酚反应生
(4)与芳香族酚的反应 氰酸酯可以与酚类化合物反应生成二芳基亚胺碳酸酯,在热与催
成亚氨基碳酸酯,在发现这种合成方法的初期,产率很低,产物纯度不高,因此很难将此法应
化剂作用下发生环三聚生成三嗪环结构。
用于氰酸酯树脂的规模化、商业化生产。但是,在适当的工艺条件下,用此反应也能制备出高
Ar-OCN+Ar'-OH
NH
纯度的芳香族氰酸酯。例如在季铵盐存在的条件下,把酚钠水溶液与高度分散在水溶液中卤化
Ar-O-C-O-
Ar'-OCN+Ar'-OH
氰反应,即可制得高纯度的氰酸酯,在季铵盐的催化和低温条件下,多烷基酚铵盐与有机溶剂中过量的卤化氰反应也可制得高纯度氰酸酯。
Ar-OCN+Ar'-OH 7.1.3 酚类化合物与碱金属氰化物的反应
OAr
加热/催化剂 {pK,(ArOH)<pK,(Ar'OH)]
将单质溴加入氰化钠或氰化钾的水溶液中,然后在叔胺(TA)存在下,将它分散人酚类化
ArO
OAr 合物的四氯化碳溶液中反应:
Br+NaCN+ArOH+TA→ArOCN+NaBr+TA·HBr 氰酸酯网络结构
098
099
聚合物基复合材料
第7章 氟酸酯树脂
7.2.2 环三聚反应及氰酸酯的固化机理
氰酸酯在热或催化剂的作用下,可以发生环三聚形成三嗪环,环三聚反应可以被酸、酚类化合物催化。
3Ar-OCN 加热/催化剂 ArO OAr 氧服酸酯 OAr■
NCO-Ar-OCN 加热/强化剂 3Ao OArT 三维氰酸酯网络
研究表明,绝对纯的芳香氰酸酯即使在加
ArO-C-0
热条件下也不会发生聚合反应。芳香氰酸酯的
结构通式为:Ar—O—C=N。由于氧原子和C
OH
氮原子的电负性,使其相邻碳原子表现出较强NH
的亲电性(Ph—O—C8+—N6—),因此,在亲
核剂作用下,氰酸酯官能团在酸或碱催化下可ArO
发生反应。
HO-
通过不同方法合成的氰酸酯,有的不含有NH
残余酚,有的含有微量的残留酚,但即使含有
HO
残留酚的氰酸酯的固化反应也非常缓慢,要使
HN
高纯度的氰酸酯单体聚合反应,必须加入催化剂提高固化反应速率。常见的催化剂有两类:
一是带有活泼氢的化合物,如单酚、水(2%~NH
6%)等;二是金属催化剂,如路易斯酸、有机金属盐等。由于氰酸酯官能团含有孤对电子和给电子π键,因此它易与金属化合物形成配合
CH 物。因此,像金属羧酸盐、ZnCl2、AICl3这样
CH
的化合物可以作为催化剂催化氰酸酯官能团的
图7—3 Loustalot等提出的制备氰酸酯树脂反应机理
三聚反应,但是这些金属盐作为催化剂时,其在氰酸酯树脂中的溶解性很差,因而它们的催
Ar-O-C=N:+TiCL
化效率很低。为了提高催化剂的催化效率,加
Ar-O-C-N:
人能溶于氰酸酯树脂中的有机金属化合物可有ArOH
效地提高氰酸酯固化反应的催化效率。在反应
Ar-O-C-N
OAr
过程中,在氰酸酯分子流动性较大的情况下,
C-OAr
金属离子首先将氰酸酯分子聚集在其周围,然后酚羟基与金属离子周围的氰酸酯亲核加成反应生成亚胺碳酸酯,继续与两个氰酸酯加成井闭环脱去一分子酚形成三嗪环。反应过程中金属OAr
过质子的转移促进闭环反应。研究人员曾提出几盐是主催化剂,酚是协同催化剂,酚的作用是通
OAr
图7—2 氰酸酯树脂聚合反应的Simon—Gillham模型
种反应机理,如图7—2~图7—4所示。
ArO
OAr
图7—4 Brownhill's提出的氰酸酯树脂缩聚成环反应机理
100
101
聚合物基复合材料
第7章 氟酸树脂
对于首面解(PTBPCN)在无便化条件下在丁酮和丙酮溶液中100它反应5后,续表
中
结
构 牌号/供应商
熔点 T.■ 介电常数 介电损耗吸湿率
/℃ /℃ (1MHz) (1MHz)
1% 制的反应量合催化剂后在10℃下,加热上h即发生了明显的三聚反应,并有少量的氰酸面数
生了水解反应。
XU-71787/
Dowchemical 2.8
0.002
1.4 7.2.3 物理性能
氰酸酯树脂的物理性能因分子结构的不同,表现出很宽广的变化范围,物理状态可以是被①树脂固化物的玻璃化转变温度。
体、晶体以及树脂状固体等,例如双酚A型氰酸酯(BCE),合成的粗品BCE单体在常温下为
表7—3 不同官能团氰酸酯树脂的熔点
淡黄色至白色颗粒状晶体,熔点为74℃左右,提纯后的BCE单体在常温下为白色粉末状结晶,
熔点/℃
名
称 熔点/℃
名
称 熔点为79℃,表7—2列出几种商品化的氰酸酯的性质。表7—3列出不同官能团氰酸酯树脂的熔点。
间苯二氰酸酯 80
2.2'—二氟氧基—1,1'—联萘149
对苯二氟酸酯 115~116
2,2'—二(4—氰氧基苯基)丙烷82
表7—2 几种商品化的氰酸酯的性质
1.3.5—三氰酸酯
102 2.2'—二氧氧基二苯基硫砜
169~170
结 称
式 牌号/供应商
熔点 T,① 介电常数 介电损耗吸湿率
4.4—二氟氧基联苯131
2.2'—二氧氧基—3,3,3',5'—四甲基二米醚107~108
1℃ /℃ (1MHz) (1MHz)
X/
CH
NCO OCN
Arocy B/Ciba-Geig 7.2.4 工艺性能
79 289 2.91 0.004
2.5 BT-2000/Mitsubishi
氰酸酯树脂具有良好的溶解性能及工艺性能,例如BCE单体在室温下易溶解于丙酮,形成CH
H,C CH
无色透明溶液,常温下放置不分层,BCE单体在80℃熔融后,具有极低的黏度(300mPa·s)。
NCO- OCN
氰酸酯树脂用于复合材料加工具有类似环氧树脂的加工性能,可以适应包括预浸料、树脂传递Arocy M/Ciba-Geig
106 252
2.75 1.4
模塑、缠绕、挤拉、压力模塑和压缩模塑等各种加工方法的要求,可以用传统的复合材料加工H1C
CH
设备加工。
CF
OCN 7.2.5 流变性能
Arocy F/Ciba-Geig 87 258 2.66 0.003
1.8 CF:
热固性树脂的流变行为主要受到两方面的影响:一方面是温度的升高导致树脂黏度的下降,
另一方面是固化反应过程中由于分子量的增加所引起黏度的增加。为了描述温度和固化反应对
NCO CH OCN
热固性树脂流变行为的影响,已有多种树脂流变模型。本书中仅介绍等温条件下和动态条件下
Arocy L/Ciba-Geig 29 252 2.98
0.005 的流变性。采用流变仪测试配制好的树脂体系。
2.4
(1)等温条件下的流变特性 温度越高,树脂黏度上升得越快,130℃时110min后黏度开
NCO OCN
始增大;140℃时60min后黏度开始增大;150℃时30min后黏度开始增大;160℃时10min后黏AroCy/Ciba-Geig
273 2.75 0.003
1.4 -
度开始增大。预聚后的氰酸酯树脂黏度受反应温度的影响很大,可见注射温度的选择对于成型
NCO 工艺是十分重要的。
OCN AroCy T-10/
(2)动态条件下的流变特性 动态条件下,氰酸酯树脂体系黏度随温度的变化:初期树脂
Ciba-Geig 192
2.80
0.004 黏度随着温度的升高而下降;固化反应开始后,树脂黏度的增加与因为温度升高所导致的黏度
1.5 CH:
CH,
的降低相抵消,因此树脂黏度下降速度减缓;固化反应所引起的黏度增加超过了因温度升高所
XU-366/Ciba-Geig
导致的黏度降低,因而树脂黏度上升。
CH
CH, RTX-366
68 350 2.64 0.001
0.7 NCO
7.2.6 氰酸酯树脂固化物的性能OCN
OCN
XU-366/Ciba-Geig 氰酸酯自聚形成的三嗪环结构的规整性好、结晶度高、交联密度较大,加上整个结构中有
Primaset PT/Lonsa
244 较多具有刚性的苯环结构,氰酸酯树脂固化物兼有高玻璃化转变温度和相对较高韧性的性能特
3.8 3.08
征。氰酸酯树脂固化物具有优异的介电性能(在25℃,1MHz下,介电常数ε=2.66~3.08、介
电损耗tanδ=1x10—3~6x10—3),在高玻璃化转变温度树脂中也是非常难得的,这可能是聚
102 氰脲酸酯网络中弱的偶极作用特征造成的。
103
聚合物基复合材料
第7章
氟酸酯树脂 (1)力学性能 繁酸酯树脂的韧性介于双马来酰亚胺和环氧树脂之间,强度和模量与二
(3)介电性能 在聚氰脲酸酯网络结构中,电负性大的氧原子和氮原子对称围绕电负性小能环氧材的氧当,例如双酚A型氰酸脂固化物的冲击强度为5.2k/㎡左右,弯曲强度为
的碳原子的结构平衡了电子吸引作用,使得偶板运动短暂,在电磁场中的储,以吸湿率能环氧方右,拉伸强度为70.3~82.7MPa,拉伸模量为2.96~3.24GPa,拉伸断裂伸长率为
和介电常数都很小。聚氰脲酸酯的另一个特征是没有强的氯键,这也使得吸湿率和介电损耗低。8.593.8%,断裂韧性(Km)值为0.62MPa·m2左右,是高性能热固性树脂中韧性较高的
航酸醋均聚物的介电常数和介电损耗都比传统的高性能树脂如环氧树脂要低,且氟酸酯均聚物一类,表7—4列出氟酸脂及其聚合物的性能,从中可见其玻璃化转变温度是比较高的。低相抵
的介电常数几乎无频率依赖性。消,因此树脂黏度下降速度减慢。固化反应所引起的黏度增加超过了因温度升高所导致的黏度
(4)黏结性能 氰酸酯树脂胶黏剂在高性能高温胶黏剂中的应用越来越形成对环氧树脂胶降低,因而树脂黏度上升,
黏剂的挑战。氰酸酷树脂胶黏剂的优点包括:与金属极好的黏结力:比环氧更优的耐湿热性能表7—4 氨酸酯树脂的性能比较
(约180℃):加工、固化范围很宽;固化过程无低分子物放出,所以黏结操作无需高压;对表面
能
环氧树脂 氰酸酯树脂
BT树脂 润湿性较好;固化无收缩现象。
性
(5)耐化学腐蚀性能 氰酸酯树脂耐化学腐蚀性能特别好,Shimp等报道称氰酸酯均聚物玻璃化转变温度/℃
110 255
310
青曲强度/MPa 6
6 6
可耐印刷线路板生产中的去脂剂、蚀刻剂、脱漆剂及其他化学品,也可耐结构复合材料遇到剥离强度/kgl/cm
的航空油、压力油和颜料脱除剂等。Shimp比较了各种树脂的耐化学腐蚀能力,发现只有室温
1.7~1.9 1.7~1.9
1.5~1.7 NaOH可侵蚀AroCyB均聚物,使树脂表面皂化,将环氧树脂与氰酸酯树脂共混可有效提高
150℃ 1.7~1.8
1.6~1.8 1.4~1.6
介电常数 4.8
4.2 4.1
耐碱性能。
介电担耗 0.02
0.003 0.002
体积电阻/Ω·cm 7.3 应用
室温 1x10
1x10■ 1x1015
150℃ 1x10#
5x10 5x10
r
Grigat 等描述了芳基氰酸酯在酸性介质中生成亚胺碳酸并重排为甲酸酯的过程。芳基氰酸酯与水的反应速率比异氰酸酯与水的反应速率要低几个数量级,但水与氰酸酯基的反应在印刷
拓展阅读
线路板(PCB)和结构复合材料的制造中仍是一个值得注意的问题。同时,氨基甲酸酯的形成
也是氰酸酯树脂热氧降解的关键。氰酸酯类聚合物的饱和吸湿此环氧树脂、BMI及缩合型聚酰7.4 发展趋势与前景
亚胺都要低,氰脲酸酯键可耐沸水数百小时。提高氰酸酯聚合物耐水性的关键是提高转化率,而提高转化率,催化剂的选择很重要。在常温下,氰酸酯单体与水基本无作用,但在催化剂存
自从1972年联邦德国Bayer公司首先将氰酸酯树脂商品化以来,短短的几十年时间内,氰
在下易形成氨基甲酸酯,含锌的催化剂尤为严重。
酸酯以其优异的电绝缘性能、极低的吸湿率、较高的耐热性、优良的尺寸稳定性、良好的力学
(2)热性能 作为应用于航空复合材料和印刷线路板(PCB)的聚合物基体,氰酸酯树脂
性能以及与环氧树脂(EP)相近的成型工艺性等,备受人们的青睐,在电子、航空航天、医疗
有很高的热稳定性,这是氰酸酯树脂最重要的特性之一,表7—5列出几种商品化的氰酸酯树脂
器材等诸多领域获得了广泛的应用,成为继环氧树脂(EP)、聚酰亚胺(PI)、双马来酰亚胺
的耐热性能,氰酸酯树脂由于树脂结构中含有热稳定性接近苯环的芳香对称三嗪环而具有较高
(BMI)之后的又一种高性能复合材料树脂基体。虽然氰酸酯具有上述优异的综合性能,但是由
的热稳定性。
于氰酸酯单体聚合后的交联密度大,加上分子中三嗪环结构高度对称,结晶度高,造成的氰酸表7—5 几种商业化氰酸酯树脂的耐热性能①
酯的固化物较脆,所以对于许多应用场合来说,仍不能满足要求。为此,人们研究了多种改性种类
玻璃化转变温度/℃ Ta/C
Tp/C 的途径,目前用热固性树脂、热塑性树脂、橡胶弹性体、含不饱和双键的化合物与氰酸酯共混,
900℃残留率/% Xu-366
192 439
以及不同结构的氰酸酯树脂单体共混或共聚等改性取得了较大的进展。482
XU-71787 244
447 463
31
B-10 257
33 7.4.1 新型氰酸酯的合成
443 468
M-10 252
443 39
在氰酸酯单体的分子结构中引入活性基团,合成带第二活性基团的氰酸酯单体,通过第二471
L-10 258
455 479
41 活性基的聚合反应,改善氰酸酯树脂的性能。Barton等合成了数种带有烯丙基的氰酸酯单体,
F-10 270
453 47
这些单体可以与BMI共聚,实现氰酸酯树脂的改性。465
BPCCE 275
441 49
461 OCN
XU-371 >350
454 99
461 NCO
PT-30 >350
457 62
462 63
2.2'—双(3—烯丙基—4—氰酸酯基)异丙烷注:Ta为失重率为5%是对应的温度;Tg为最大失重率对应的温度。
1—烯丙基—2—氰酸酯基苯在氰酸酯单体结构中引人具有功能性的基团或链段,赋予氰酸酯树脂功能性,例如在氰酸
酯单体中引入苯基磷结构,可以赋予氰酸酯树脂优异的耐热和阻燃性能,如图7—6所示,其性
105
聚合物基复合材料
第7章 ■酸酯树脂
能随苯基磷与三嗪环的比例不同而变化。
化反应。而更为少量的环氧树脂也能促进氧酸酯树脂的因化反应。由此可见,氧酸酯改性环氧树脂是化学改性。CE可与EP发生共聚合反应,生成氧联环、异氧酸酯环、唑烧环及三嗪环答,CE/EP改性体系既能形成大量的三嗪环,保留CE固有的性能优点,又能与EP共固化而形成交联网络,提高了材料的力学性能;树脂固化物中含有大量的醚键,因此具有较高的韧性和冲击性能,通常用作高性能印制电路板的基体树脂。
表7—6中,A为氰酸酯预聚体与环氧树脂组成的二元体系;B为氰酸酯先与少量环氧树脂共CH
聚后,再与环氧树脂共混形成的改性体系,可以发现改性体系的硬度和模量略有降低,但强度
提高显著。另一方面,除了断裂伸长率略有不同外,两种改性工艺的力学性能基本相当,这体CH3
现了良好的改性工艺。
表7—6 CE/EP改性体系CH
OH 性
能 氰酸酯树脂
A B
CH
CH 巴氏硬度
48 38
40 拉伸强度/MPa
50 81.6
72.4 HO
新裂伸长率/% 1.42
5.69 4.30
弯曲强度/MPa 80.9
147.7 149.5
(CH)N
CNBr 奇曲模量/GPa
4.6 2.9
2.8
0.02- THF
OPh (2)与BMI共聚改性 CE/BMI共混或共聚改性是一个较为活跃的研究领域,BMI改性CE
CH3 最直接的一个方法就是BMI与CE熔融混合得到均相的共混体系,在较低的温度下即可发生共
CH 聚,CE官能团(—OCN)与BMI的马来酰亚胺环上的不饱和双键上的活泼氢发生反应,得到
BT树脂。BT树脂的玻璃化转变温度达250℃以上,具有较低的介电常数和介电损耗因数、优良的抗冲击性能,通常用作高性能印制电路板的基体树脂。BT树脂体系的玻璃化转变温度随BMI含量的增加而提高,但力学性能及工艺性能等变差。
(3)BMI/EP/CE三元改性体系 EP/CE改性体系降低了CE原有的模量、耐热性及耐化学
CH 腐蚀性能;BMI/CE改性体系的增韧效果不太明显,且其工艺性能较差,成本较高。因此许多
R: 研究者应用这三种树脂共混,以期得到性能更佳的树脂体系。BMI/EP/CE三元体系中,EP与
CH, CE的共聚结构与BMI形成互穿网络结构(IPN),使体系的工艺性和韧性比二元体系有了较大
的提高。 Bobert E.Hefne 等对三元改性体系进行深入研究,其三元改性体系浇注体的性能见表7—7。
表7—7 BMI/EP/CE三元改性体系浇注体的性能OPh
性
能 CE
A B
图7—6 氰基苯基磷和交联的苯基磷—三嗪聚合物的合成
83
89.2 拉伸强度/MPa
50
断裂伸长率/% 1.42
9.63 9.48
7.4.2 共混改性155.0
弯曲强度/MPa 80.9
136.2
弯曲模量/GPa 4.6
3.0 3.1
(1)与环氧树脂(EP)共聚改性 环氧树脂是一类综合性能优良的树脂,在复合材料中得
HDT/C
192 183
254
到了广泛的应用。但是由于环氧树脂基体中含有大量的固化反应生成的羟基等极性基团,环氧树脂固化物的吸湿率较高,使其复合材料在湿热环境下力学性能显著下降。利用氰酸酯树脂改
其中A为CE先与BMI共聚,再与EP共混得到的三元体系;B为CE先与EP共聚,再与
性(固化)的环氧树脂其固化分子中不含羟基、氨基等极性基团。因此,固化物耐湿热性能好。
BMI共聚得到的三元体系。表7—7中数据表明,三元改性体系具有更好的增韧效果,弯曲模量
另外,固化物中含有五元含哪唑啉杂环和六元三嗪环结构,因此其固化物有较好的耐热性;固
有所提高,综合性能明显优于二元体系。另外,A、B两种改性工艺得到的树脂体系性能基本相
化物分子中含有大量的—C—O—C—醚键,故又具有较好的韧性。
当,也说明了三元改性体系具有良好的工艺特性。
在无催化剂和固化剂条件下,无论是氰酸酯还是环氧树脂都很难进行固化反应,但是,当
(4)热塑性树脂改性CE CE可与许多非晶态的热塑性树脂共混。热塑性塑料所占的质量
氨酸酯与环氧树脂混合时,两者能互相催化固化反应,少量的氰酸酯树脂能促进环氧树脂的固
分数为25%~60%(视性能要求而定)。所用的热塑性塑料主要为玻璃化转变温度较高和力学性
106 能比较优良的树脂,如聚碳酸酯(PC)、聚砜(PSU)、聚醚砜(PES)、聚醚酰、聚醚酰亚胺
107
聚合物基复合材料
第7章 氟酸腿树脂
(PE)等,上述种霸可溶于俗减的氟酸器器中,因此可用热格法或格融挤出法制备的树脂浇注体的性能见表7—10。从表中可以看出,CE/MMA体系的综合性能较好,力学性能较
纯CE有了较大的提高,韧性也得到明显的改善。
表 7-10 CE/不饱和双键化合物改性体系的性能
并发生分相成为变力时产生的微裂纹的扩展,提高了材料的初性,随若热塑性塑料用的性
能 CE
CE/苯乙烯 CE/MMA
CE/丙烯酸丁酶
拉仲强度/MPa 50
50.3 85.1
63.1 分能与热原性塑料最终形成半互穿网络结构,从而得到一种高力学性能、高使用温度
艺性及耐热、耐湿性能等。表7—8为几种CE/热塑性树脂共混体系的性能。我脂材第一化学结构的材料体系。这既提高了氰酸酯树脂的初性,又改善了热塑性塑料的工
断裂伸长率/% 1.42
0.5 3.1
1.8
弯曲强度/MPa
80.9 127.9
169.9 112
热塑性树脂的加入会使CE的耐热性有不同程度的下降。此外,由于热塑性塑料的分子量较冲击强度/(kJ/㎡)
5.2 5.1
6.5 7.2
寿曲模量/GPa 4.5
11 3.9
3.4 大,共混树脂的黏度增大,工艺性变差。因此,在使用热塑性塑料改性氰酸酯树脂时,需要考
马丁耐热温度/℃ 一
158 135
112 虑热塑性塑料对氨酸酯韧性、耐热性和工艺性能的影响,以便获得综合性能相对较佳的体系。
表7—8 热塑性树脂/双酚A氨酸酯(1+1)semi—iPN结构树脂的性能
拉伸强度 拉仲模量
断裂伸长率 热失重温度
玻璃化转变温度 热塑性树脂
/MPa /GPa
1% /℃
/℃
聚酯/碳酸酯共聚物 84.5
2.14 17.6
-
聚碳酸酯 84.7
2.06 17.3
400 195
聚■ 73.0
2.05 12.7
350 185
聚对苯二甲酸乙二醇酯76.5
2.44 12.5
■■■ 71.7
2.34 9.6
聚型酰亚■ 76.0
2.44 12.5
期码
(5)橡胶弹性体改性氰酸酯树脂 P.C.Yang、D.M.Pickelman和E.P.Woo等对橡胶增韧 氰酸酯树脂提出了一种核—壳结构增韧机理,他们所使用的氰酸酯树脂是一种牌号为Xu71787的芳杂环结构的氰酸酯树脂。核为橡胶,壳为氰酸酯树脂固化物。材料在受到外力的作用下发生变形时,核—壳结构发生位移而产生空穴,空穴吸收能量,起到了增韧的作用。使用橡胶增韧氰酸酯树脂,可在较低的温度(80℃)下与氰酸酯树脂共混。橡胶的加入也不像热塑性塑料那样会对树脂的黏度产生较大的影响。但是由于橡胶的耐热性问题,固化条件对改性体系的性能有
S作第报学热必与 较大的影响。橡胶改性氰酸酯树脂体系的后处理温度不宜过高,因为高温会使橡胶老化。
CE最常用的橡胶增韧剂为端羧基丁腈橡胶(CTBN),橡胶/CE体系浇注体的性能见表7—9。日
但是由于橡胶的耐热性问题,固化条件对改性体系的性能有较大的影响,橡胶改性CE体系的后处理温度不宜过高。
表7—9 橡胶/CE体系浇注体的性能
性
能 橡胶含量
助人数
0 0.5%
5.0% 玻璃化转变温度/℃
10.0% 吸水率/%
250 253
0.7 0.76
254 254
弯曲强度/MPa 121
0.95 弯曲模量/GPa
117 0.93
应变/% 3.3
3.1 112
101
Cm/(kJ/㎡) 4.0
5.0 2.7
2.4 0.07
6.2 7.5
Ki/MPa·m/ 0.20
0.552 0.837
0.32 0.63
1.107 1.118
(6)不饱和双键的化合物改性CE 在催化剂的作用下,CE可与苯乙烯、丙烯酸丁酯、甲基丙烯酸甲酯(MMA)、不饱和聚酯等不饱和双键的化合物共聚形成改性体系。一些改性体系
108
109
第8章 聚酰亚胺树脂
第8章 聚酰亚胺树脂
8.1 概论 20
0 360 380 400 420 440 460 480
500 聚酰亚胺是指主链上含有酰亚胺环的一类聚合物,这类聚合物早在1908年就有报道,其一
温度/℃ 般结构为:
图8—1 不同类型聚酰亚胺的热稳定性
双马来酰亚胺;→热塑性:乙炔基封端:缩聚型
8.1.2
合成 聚酰亚胺树脂可分成缩聚型、加成型和热塑性三种类型。
聚酰亚胺的合成方法可以分为两大类,第一类是在聚合过程中,或在大分子反应中形成酰8.1.1 性能
亚胺环;第二类是以含有酰亚胺环的单体聚合成聚酰亚胺。(1)聚合过程中或在大分子反应中形成酰亚胺
①全芳香聚酰亚胺按热重分析,其开始分解温度一般都在500℃左右。由联苯二酐和对苯①由二酐和二胺反应形成聚酰亚胺
二胺合成的聚酰亚胺,热分解温度达到600℃,是迄今为止聚合物中热稳定性最高的树脂之一.②聚酰亚胺可耐极低温,在—269℃的液氮中仍不会脆裂。
③聚酰亚胺具有很好的力学性能,未填充塑料的拉伸强度都在100MPa以上。均苯型聚酰亚胺的薄膜(Kapton)为170MPa,而联苯型聚酰亚胺(UpilexS)达到400MPa。
④一些聚酰亚胺品种不溶于有机溶剂,对稀酸稳定,一般的品种不耐水解,可以根据这一
此方法是合成聚酰亚胺最普遍的方法,反应分为两步:第一步是将二酐和二胺在非质子极
特点利用碱性水解回收原料二酐和二胺。例如对于(Kapton)薄膜,其回收率可达80%~90%。
性溶剂,如二甲基甲酰胺(DMF)、二甲基乙酰胺(DMAc)、N—甲基吡咯烷酮(NMP),或四
⑤聚酰亚胺的热膨胀系数在(2~3)x10—5/℃,联苯型可达10—6/℃,个别品种可达10—7/℃.
氢呋喃和甲醇混合溶剂中进行低温溶液缩聚,获得聚酰胺酸溶液,去除溶剂后,再经高温处理
⑥聚酰亚胺具有很高的耐辐射性能,其薄膜在5x10°rad剂量辐照后,强度仍保持86%。
形成聚酰亚胺。聚酰胺酸也可用化学脱水剂,一般用乙酸酐为脱水剂、叔胺类(吡啶、三乙胺
一种聚酰亚胺纤维经1x1010rad快电子辐照后其强度保持率为90%。
等)为催化剂,在室温下酰亚胺化而获得聚酰亚胺。
⑦聚酰亚胺具有很好的介电性能,介电常数为3.4左右,引入氟、或将空气以纳米尺寸分
二酐和二胺也可用一步法形成聚酰亚胺,即将两种单体在高沸点溶剂中加热至150~250℃
散在聚酰亚胺中,介电常数可降到2.5左右。介电损耗为10—3,介电强度为100~300kV/mm,
而获得聚酰亚胺,所用的溶剂可以是酚类,如甲酚、对氯苯酚、邻二氯苯或1,2,4—三氯代苯等。
体积电阻为107Ω·cm。这些性能在宽广的温度范围和频率范围内仍能保持在较高水平。
酚类溶剂的优点是可以溶解多种聚酰亚胺,可得到高分子量的聚合物。其他溶剂往往会使聚合
⑧聚酰亚胺为自熄性聚合物,发烟率低。
物在分子量增长到一定程度后就从溶液中沉淀出来。
⑨聚酰亚胺在极高的真空下放气量很少。
②由四元酸和二元胺反应生成聚酰亚胺
⑩聚酰亚胺无毒,可用来制造餐具和医用器具,并经得起数千次消毒。一些聚酰亚胺还具有很好的生物相容性,例如,在血液相容性试验中为非溶血性,体外细胞毒性试验为无毒。
COOH
COOH
严格来讲,只有加聚型的聚酰亚胺是热固性的树脂,因为加聚型的聚酰亚胺是分子量较小的酰亚胺化的低聚物,通过活性端基进行交联固化,形成网状结构。固化树脂具有较高的交联
该反应在高沸点溶剂中进行,先由四酸和二胺形成盐,然后在高温下脱水形成聚酰亚胺,
密度,因此具有较大的脆性。缩聚型聚酰亚胺其行为像热固性树脂,树脂固化物是不溶不熔的。
也可以是四酸在高温下,如150℃以上脱水成酐,再与二胺反应。
聚酰亚胺作为先进复合材料基体应用的主要原因在于其能在250℃以上长期使用,这一点即
芳香四酸通常在100℃以上就会脱水成酐,所以当以四酸为原料时,应保证四酸中没有二
使最好的多官能团环氧树脂也不能达到,不同类型聚酰亚胺的热稳定性如图8—1所示。
酐,也没有水分,否则会由于四酸和二胺达不到等摩尔比而得不到高分子量的聚合物。
另外,有两个主要原因阻碍了经典聚酰亚胺作为结构复合材料基体的广泛应用。第一,交
③由四酸的二元酯和二胺反应获得聚酰亚胺
联密度高、分子链刚性大的聚酰亚胺固有的脆性,导致了复合材料耐损伤性差以及热冲击时基
COOH
体树脂易开裂,这种开裂则使吸湿性增加以及冷热交替时易变形;第二,聚酰亚胺的可加工性差,聚酰亚胺的加工一般需要在高温(250~300℃)下进行,缩聚型聚酰亚胺还需要较高的压力。
COOH
110 8 8
质量保留率(2h)/%
聚合物基复合材料
该方法是首先将二肝在醇中回流酯化,得到二般二酯,冷却后加入二胺和第三组分,例第8章 聚酰亚胺树脂
二无酸的单作溶液作为浸渍料,涂覆在碳纤维或玻璃纤维上,经过热处理后再热压成题,CH
成以热塑性或热固性聚酰亚胺为基体的复合材料。
④由二酐和二异氰酸酯反应获得聚酰亚胺
盐或双硫酚盐进行亲核取代反应可以获得聚醚酰亚肢或聚硫醺酰亚胺。由于苯环上的卤素或硝基可以被酰亚胺基团活化,以双卤代酞酰亚胺或双硝基酞酰亚胺与双酚
此方法是大幅度降低聚酰亚胺成本的重要途径之一,尤其在用邻二甲苯经氯代、氧化并分离的高纯度的3—氯代苯酐和4氯代苯酐的合成路线开发成功之后,其意义更为突出。
分离后异构体的纯度达99%以上:气相氧化收率为70%;3—氯代苯酐/4—氯代苯酐=34/66;液相氧化收率为80%,3—氯代苯酐/4—氯代苯酐=45/55。
该反应的优点是不产生水分,只产生容易逸出的二氧化碳。但由于异氰酸酯十分活泼,可该反应在本质上与用亲核取代反应合成聚醚砜或聚醚酮相同,不同的是酰亚胺环在碱性介
以发生许多副反应,如二聚、三聚成环,还可以通过C—N双键聚合形成尼龙。异氰酸酯和空质中,尤其在较高温度下会发生分解。因此往往难以得到高分子量的聚合物,解决的办法应有
气中的水分接触容易发生水解,使纯度降低,因此常使反应复杂化。以下几点:a.控制反应温度在尽可能低的范围,例如150℃以下;b.选择溶解性较好的结构,
⑤邻位二碘代芳香化合物和一氧化碳在钯催化下与二胺反应转化为酰亚胺避免聚合物过早地在反应介质中沉淀出来;c.严格控制体系的含水量,例如在100mg/L以下;
0
0 d.如果可能,应选用活性更大的离去基团,和使离去基团处于对活性更有利的3—位。
L-PdCl: ②用酰亚胺交换反应获得聚酰亚胺
此反应的特点是收率高,副反应少。为了得到高分子量,应使产生的聚合物保持在溶液中+RNH2
以使分子量继续得到增长。
⑥由酯基或酰氨基的邻位碘代物在钯催化下与一氧化碳反应得到聚酰亚胺COOR
COOR CONR
O Pd(O)/CO
在20世纪60年代就有专利报道以均苯四酰亚胺在极性溶剂中和二胺反应,室温下可得聚0.6MPa,120℃
P-1
酰胺酰胺,加热脱氨得聚酰亚胺。有人采用R=—OCOEt这种较为活泼的基团获得高分子量的聚合物。
CONR CONR
R ③由带酰亚胺环的二卤化物与二硼酸化合物在钯催化剂作用下缩聚得到聚酰亚胺
Pd(0)/CO
R 0.6MPa,120℃
Pd-1
(HO)2B B(OH)2
Br
0
该方法还得不到高分子量的聚合物,可能的原因之一是脱出的醇或胺仍可以与钯配合物作用产生邻位二酯或二酰胺。
Pd(PPhy)4 O
R
R
⑦由二酐的二氰基亚甲基衍生物与二胺在低温下反应生成聚酰亚胺
甲苯Na,CO3水溶液
NC CN
为了增加溶解度,R为烷基。反应在回流条件下48h完成。④由四酰二亚胺的碱金属化合物和二卤代物反应获得聚酰亚胺
0
0 0
该反应放出丙二腈,以均苯二酐的衍生物与二苯醚二胺在NMP中反应,室温下24h,获得
XN
酰亚胺化达75%的均相溶液,继续放置则析出沉淀。固态下120℃经20h酰亚胺化可基本完成。
X=H,K:R=各种芳香和脂肪基团
(2)用带酰亚胺环的单体缩聚获得聚酰亚胺
0
①以双卤代酞酸亚胺或双硝基酞酰亚胺和双酚或双硫酚的碱金属化合物间的亲核取代聚合
三正丁胺>吡啶>碳酸钾。当X为K时反应速率和收率都可得到提高。
合成聚酰亚胺 当X为H时反应在碳酸钾和叔胺催化下进行,催化剂对收率的影响有下列关系:三乙胺>
8.1.3 聚酰胺酸的合成和酰亚胺化
112 最常用的聚酰亚胺的合成方法是由二酐和二胺在非质子极性溶剂中先形成聚酰胺酸,然后
113
聚合物基复合材料
再用热或化学方法脱术成环,转化为聚酰亚胺,其主要反应过程如图8—2所示。
第8章 聚酰亚胺树脂
表8—2 含水溶剂对聚酰胺酸的黏度及聚酰亚胺性能的影响聚酰胺酸胶液性能
二酐/二胺 DMAc含水量/%
聚酰亚胺薄膜性能
PMDA/ODA
7m/(dL/g) 7m/(dL/g)
0.1 6.60
拉神强度/MPa
PMDA/ODA
1.0 3.30
一 伸长率/%
PMDA/ODA 5
139 66
一
PMDA/ODA 1.55
-
10 一
一
PMDA/ODA 1.12
20 -
-
PMDA/ODA 30
0.96 131
66
一
BPDA/ODA 0.1
0.46 134
43
-
BPDA/ODA 2.05
不能成膜 5
1.37 一
BPDA/ODA 10
124 20
-
BPDA/ODA 15
0.93
- 115
23
图8—2 二酐和二胺合成聚酰亚胺的主要反应BPDA/ODA
0.85 114
17 HOO
- 25
0.73 112
BPDA/ODA 30
- 11
非均相 81
8
ODPA/ODA 0.1
(1)聚酸胶酸的合成 聚酰胺酸是由二酐和二胶在非质子拔性溶剂,如N,N二甲基ODPA/ODA
0.97 1.09
10 0.40
107 19
ODPA/ODA 15
0.36 1.03
121
酸(DMF)—10℃~室温)下反应得到,由于二酐容易被空气或溶剂中的水分水解,得到的ODPA/ODA
1.02 15
20 0.25
110 9
ODPA/ODA 25
1.05 101
中于低截在低温下不能与二酸反应生成酰胺,从而影响到聚酰胺酸的分子量,为了保证获得ODPA/ODA
0.22 0.99
11
30 非均相
106 10
分子量的聚酰能酸,在使用菌应将反应器和溶剂干燥,三肝应在使用前妥善保存避免被空气TDPA/ODA
0.1 1.12
的水分水解。对于对水解特别敏感的二酐,如均苯二酐,最好使用刚脱过水(例如升华)的二HQDPA/ODA
5 0.85
1.09 121
15
HQDPA/ODA 15
1.02 109
6
肝。反应时应将二酐以固态加入二胺的溶液中,同时开始搅拌,必要时还要外加冷却。然而在0.58
1.03 112
HQDPA/ODA 20
0.44 12
实际应用时,分子量太高的聚酰胺酸由于溶液黏度太大而不便于加工,难以得到薄而均匀的薄TDPA/ODA
0.5 1.12
96 8
非均相
膜,此外还经常发生在二酐溶解之前体系就变得十分黏稠,使反应难以顺利进行,或者在溶液HQDPA/ODA
0.1 1.02
1.15 HQDPA/ODA
5 0.91
104 12
中形成聚合物的团块,影响后期的加工,所以可以根据需要将聚酰胺酸的分子量控制在一定的HQDPA/ODA
10 1.11
105 17
范围。为了达到这个目的,最好的办法是使用含有一定水分的溶剂,也就是让二酐水解掉一部分HQDPA/ODA
0.83 1.01
107 15
0.69 23
1.01 12
来控制所生产的聚酰胺酸的分子量。目前在生产过程中采用多加或少加二酐来调节分子量的办法(2)二酐和二胺的活性 形成聚酰胺酸的反应是可逆的。正向反应被认为是在二酐和二胺之
是不可取的,因为这样会破坏二酐和二胺的等摩尔比,造成最终产物聚酰亚胺分子量的降低,从间形成了传荷配合物。由于酐基中一个羰基碳原子受到亲核进攻,这种酰化反应在非质子极性溶
而影响了产品的性能。由酐水解生成的邻位二酸端基在聚酰胺酸加热环化时仍然可以脱水成酐井剂中室温下的平衡常数达到105L/mol,因此很容易获得高分子量的聚酰胺酸。该反应的平衡常数
重新和端氨基反应,只要单体保持等摩尔比,聚酰亚胺的分子量仍然可以增长到所需要的程度,决定于胺的碱性或给电性以及二酐的亲电性。动力学研究表明,对于不同的二酐其酰化能力可相
表8—1是均苯二酐(PMDA)或二苯醚二胺(ODA)过量对所制得的薄膜力学性能的影响。表8—2差100倍,而对于不同的二胺其反应能力则可相差105倍,所以带有吸电子基团,如—CO一、
是将各种二酐和二胺在含水量很高的溶剂中缩聚,所得到的聚酰胺酸热环化后获得的聚酰亚胺的—SO2一、炔基及含氟基团的二胺,尤其当这些基团处于氨基的邻位、对位时,在通常的低温溶液
力学性能。由表8—1可见,由于水分的存在使聚酰胺酸分子量在一定程度内的降低并不会明显影缩聚中难以获得高分子量的聚酰胺酸。然而迄今为止还未发现二酐因为带有给电子基团而得不到
响聚酰亚胺的力学性能。足够分子量的聚酰胺酸的情况。
表8—1 在PMDA或ODA过量时所制得的薄膜的力学性能对于二酐,其羰基的电子亲和性(EA)越大,酐的电子接受能力越大,酰化速度也越高,它
过量分数/% PMDA
ODA 可以由极谱还原数据得到,也可以由分子轨道法算得。
拉伸强度/MPa 伸长率/%
拉伸强度/MPa 仲长率/%
8.1.4 聚酰胺酸的热环化
0
0.1 160
152 49
0.2 130
130 27
在由聚酰胺酸以热处理获得聚酰亚胺的过程中,除了脱水环化外还有其他反应,如聚酰胺酸61
0.5 37
130 131
37 的解离、端基重合和交联等。
1.0 29
2.0 33
最常用的测定酰亚胺化程度的方法是使用红外光谱。表8—3列出了研究酰亚胺化过程最常用125
140 35
130 153
62
5.0 111
33 的一些基团的波数:1780cm—3和1380cm—3是确定酰亚胺化程度最常使用的波数。除了红外光谱
133 58
29 106
31 以外,其他测定酰亚胺化程度的方法还有环化的热效应、介电损耗和机械损耗、核磁共振等。测
定环化时放出的水分和滴定尚未环化的羧基都是可以采用的方法。
115
表8—3 展亚胺及有关化合物的红外吸收光谱
聚合物基复合材料
度 来
第8章 聚酰亚胺树脂强
表8—4 BTDA和各种芳香二胺合成的聚酰亚胺的T2(-程/
5 二胺结构
T./℃ C-O不对称伸展
项
日 1%
C-O对称仲展 CH2
二肢结构 T./℃
1780
C-N伸展 232
5
芳香酸亚胺
1720
1380 1CH
C-O弯曲 284
725
亚氨基内酚 257
5
1750~1820 m
亚氨基内酶 283
异酸亚胺 1700
亚氨基内面
SA
921~934
277 m
COOH和 NH: 300
2900~3200 5
C-O(COOH) 酸酸酸
1710 C-O(CONH)
1660酰胺1 5
320 O
m C-NH
300 1550酰胺
CH2 m
C-O 320
1820 s
C-0 278
酐 1780
C-O
s
720
单体的化学结构对缩聚型聚酰亚胺热氧化稳定性有较大的影响。W
NH2对称结构(vs)
①对苯二胺与不同的二酐合成的聚酰亚胺,热氧化稳定性的次序如下:均苯四甲酸二酐>胺
3200两个谱带
NH2不对称结构(vas)
3,3',4.4'—二苯甲酮四甲酸二酐>1,3—二(3,4—二羧基苯)六氟丙烷二酐>1,4,5,8—萘四甲酸二酐。5
苯环的振动 ②均苯四甲酸二酐与不同的二胺合成的聚酰亚胺,热氧化稳定性的次序如下:对苯二胺>间
苯环 1500
位对苯二胺>1,5—二氨基萘≥4,4'—二氨基联苯>1,4—二氨基葱>1,6—二氨基芘,由上述热氧化稳定性次序可知,二胺中的稠环数增加,热氧化稳定性降低。
8.2 缩聚型聚酰亚胺树脂③在二胺中的环取代降低热氧化稳定性。
④用H2N—C6H4—X—C6H4—NH2结构的二胺合成聚酰亚胺时,热氧化稳定性有如下次缩聚型聚酰亚胺树脂的主要原料是芳香二酐和芳香二胺。缩聚型聚酰亚胺树脂的合成一般分
两步进行。首先,二酐和二胺室温下在极性溶剂(如二甲基甲酰胺、二甲基乙酰胺或N—甲基吡咯序:X=单键>S>SO2>CH2>CO>SO>O。
烷酮)中反应,生成可溶的聚酰胺酸预聚物。然后,通过加热或化学处理完成环化。反应方程式8.3 加聚型聚酰亚胺树脂
如下:
0 0
O
0 加聚型聚酰亚胺是指端基带有不饱和基团的低分子量聚酰亚胺,如双马来酰亚胺、降冰片烯
NH-C
C-OH 封端酰亚胺、乙炔封端酰亚胺等。成型加工时通过不饱和端基进行固化,固化过程中没有挥发性
O Ar O+H +2nH0
物质放出,有利于复合材料的成型加工,所以加聚型聚酰亚胺被广泛用于制造复合材料。HO-C
0 0
0 8.3.1 双马来酰亚胺树脂
0
二酐 二胺
聚酰胺酸
在聚酰胺酸合成时,先将二胺和溶剂加入反应釜中,再加入干燥的固体二酐。其中单体的纯双马来酰亚胺(BMI)树脂是指用双马来酰亚胺(bismaleimide,BMI)制备的树脂总称。
度、原料配比、溶液浓度和溶剂种类对聚酰胺酸的分子量有很大的影响。由于聚酰胺酸不稳定,BMI树脂具有良好的耐高温、耐辐射、耐湿热、模量高、吸湿率低和热膨胀系数小等优良特性。
必须在干燥和冷冻的条件下保存。聚酰胺酸的环化是在高温下进行的,由于聚酰胺酸的熔点接近为此,各国对BMI树脂的研究开发和应用非常重视,至今已开发出一系列性能优异的BMI树脂,
并广泛用于航空、航天和电子电气领域。环化反应的温度,故沉析作用极大地影响了树脂的流动性,因此不能用于模压和层压工艺。且聚
20世纪60年代末期,法国罗纳—普朗克公司首先研制出M—33 BMI树脂及其复合材料,并很酰胺酸在环化时要放出小分子挥发物,使制品中孔隙率增加,所以缩聚型聚酰亚胺树脂极少用作
快实现了工业化。从此,BMI树脂开始引起了越来越多人的重视。BMI树脂具有与典型的热固性为复合材料的基体树脂,一般用于制造薄膜和涂料。选用不同的原料单体可以制备不同性能的果
树脂相似的流动性和可模塑性,可用与环氧树脂类同的一般方法进行加工成型,因此BMI树脂得酰亚胺,表8—4列出了3.3,4.4—二苯甲酮四酸二酐(BTDA)和各种芳香二胺合成的聚酰亚装
到了迅速发展。 的Tg。
我国于20世纪70年代初开始BMI树脂的研究工作,当时主要应用于电器绝缘材料、砂轮黏合剂、橡胶交联剂及塑料添加剂等方面。80年代后,我国开始了对先进复合材料BMI树脂基体的
117 116
聚合物基复合材料
研究,并取得了较多的科研成果,且有的成果已商品化。定,并数请了段生的定主更件的的标法、改要期脂相性、提高制脂电作维并均氧
第8章 聚酰亚胺树脂
BMI树脂的固化产物是不溶不熔的,刚性和脆性都较大。具有相当高的密度(1.35~1.4#/cm),T。为250~300℃,断裂伸长率低于2%,BM1树脂的吸混率与环氧材脂相当(质量分数
化稳定性,以及改善树脂的加工性。
为4%~5%),但是吸湿饱和比环氧树脂快。表8—6列出了BMI树脂的性能。
双马来酰亚胺的一般结构如下:
表8—6 BMI树脂的性能
性能
最高值 BMI树脂牌号
T,/C 干态
400 性能
最高值 BMI树脂牌号
Kerimid FE70003 拉仲断裂延伸率/%
湿态 297 Ciba-Geigy XU-295
拉伸强度/MPa 干态(25℃)
2.9 Narmco 5245C
R为—CH—,—O—,—SO,或其他基团
06
双马米酰亚胺(EMI)树脂是以马来酸酐和二元胶为主要原料,经缩聚反应得到,反应方程
干态(25℃) Technochemie H795
干态(177℃) 3.3 Hysol EA9102
湿态(25℃) 88 Ciba-Geigy XU-295
断裂韧性/(J/㎡)210 Ciba-Geigy XU-295
式如下: 马来酰亚胺可均聚,也可与各种单体如乙烯基化合物、烯丙基化合物及苯乙烯类化合物以游
离基机理进行共聚,还可进行阴离子聚合。
BMI树脂 可与适当的双烯进行Diels—Alder反应,与烯丙基型烯烃的双键进行ene—反应,与伯
双马来酰亚脏树脂具有与环氧树脂类似的加工性能,而其耐热性和耐辐射性优于环氧树脂,
胺、仲胺及C—H酸性化合物在碱存在下的Michael—加成反应,同氰酸酯、异氰酸酯和环氧化合物
而且也克服了缩聚型聚酰亚胺树脂成型温度高、成型压力大的缺点。的加成反应等。许多反应研制出大量以BMI为基体的树脂商品。
BMI的均聚物由于太脆,用处有限,为了能使其作为基体树脂用在先进复合材料,必须加入
8.3.L.1 BMI单体的性能BMI单体一般为结晶固体,芳香族BMI具有较高的熔点,脂肪族BMI具有较低的熔点。从
活性稀释剂和共聚单体等以提高固化树脂的韧性及改善树脂的加工性能。8.3.1.3 BMI树脂的合成
BMI树脂的工艺性能角度,希望BMI具有较低的熔点。表8—5列出了几种常见BMI单体的熔点。目前BMI合成方法,根据催化剂与反应介质不同,可分为以下三种。
大部分BMI单体不溶于丙酮、乙醇等有机溶剂,只能溶于强极性的二甲基甲酰胺(DMF)、①以二甲基甲酰胺(DMF)强极性溶剂为反应介质,以乙酸钠为催化剂,乙酸酐为水吸收
N—甲基吡咯烷酮(NMP)等溶剂.BMI单体可通过其分子双键端基与二元胺、酰胺、酰肼、疏剂,在90℃左右进行脱水反应。其特点是中间产物双马来酰亚胺酸(BMIA)溶于溶剂中,反应
基、氰脲酸和羟基等含活泼氢的化合物进行加成反应;也可以与环氧树脂、含不饱和双键的化合体系始终处于均相,有利于反应进行;但溶剂毒性大,价格高。
物(如烯丙基、乙烯基类化合物)反应;在催化剂或热作用下也可以发生自聚反应。②以丙酮为溶剂,乙酸镍为催化剂,乙酸酐为脱水剂,在回流条件下进行。其特点是中间产
表8—5 几种常见BMI单体的熔点物BMIA从溶剂中成固体析出,反应不易均匀;但催化剂选择性好,副产物少,溶剂价格便宜,
毒性低。 R
熔点/℃ R
熔点/℃ ③不加催化剂,采用热脱水闭环法,用强极性高沸点溶剂,如DMF,在回流状态下反应,
-CH- 156~158
>340 它的特点是成本低、三废排放少。
(CH:方 190~192
8.3.1.4 BMI树脂的改性(CH
171 307~309
BMI树脂虽然具有优良的耐热性能和力学性能,但是BMI树脂熔点高、溶解性差、成型温度
(H)
137~138 高和固化物脆性大等缺点,阻碍了它的应用和发展。关于BMI树脂的改性研究有较多的报道,目
(CH) 113~118
前的研究方向主要是增韧,其目标是更优的耐湿热性能和复合材料的冲后压缩性能,以及具有好
(CH2) 111~113
CH 172~174
的可加工性能和更长的使用期。各种改性剂改性后的BMI树脂具有高的力学性能,可满足应用
(CH 110~112
要求。
CH,-C(CH1):-CH,- 70~130
307~309 文献报道的BMI树脂的改性方法较多,主要的改性方法有如下几种:用烯丙基化合物共聚改
251~253 性;用芳香二胺化合物扩链改性;用环氧树脂改性;用热塑性树脂增韧改性;用氰酸酯树脂改性。
180~181 (1)烯丙基化合物共聚改性BMI 利用BMI双键与其他活性基团化合物反应来改性BMI树脂
198~201 起始于20世纪80年代,这标志着第三代BMI的诞生,其中烯丙基化合物改性BMI树脂是最为成
154~156 功的一例。烯丙基化合物与BMI单体的预聚物稳定、易溶、黏附性好、固化物坚韧、耐湿热,并
8.3.1.2 BMI固化物的性能具有良好的电性能和力学性能等,适合用作先进复合材料基体树脂。
烯丙基化合物与BMI单体的固化反应机理比较复杂,一般认为是马来酰亚胺环的双键与烯丙
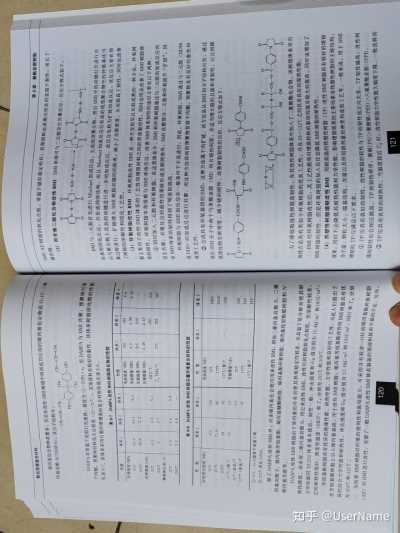
BMI树脂的突出优点是其双键的高活性,是由两个相邻的拉电子羰基的作用使双键高度缺电基在较低温度下首先进行烯加成反应生成1:1中间体,而后在较高温度下马来酰亚胺环中的双键
子,即使没有催化剂存在,在热作用下也可发生聚合。与中间体进行 Diels—Alder 加成反应和异构化反应生成高度交联的韧性树脂。
118 119
T0
第8章 聚酰亚胶树脂
聚合物基复合材料 始丙基化合指种类繁多,但是在改性BND树脂中得到成功应用的始丙基化合物是0.0'二角
BN/AE树脂的软化点低、常温下储存稳定性好,其预浸料也表现出优良的室温下黏性,延长了
丙基双酚A(DABPA),其分子结构为:
储存期。
CH-CH-CH CH CH1-CH-CH:
(2)用芳香二胺化合物改性BMI BMI单体可与二元胺发生共聚反应,反应方程式如下:
HO-
HO
CH DABPA在常温下是棕红色液体,黏度为12~20Pa·s,用DABPA与BM共聚,预聚体可溶
于丙酮,预聚体的软化点比较低(20~30℃),其预浸料有较好的黏性。该体系树脂固化物的性能BMI与二元胺首先进行Miachael加成反应,生成线型聚合物,然后BMI中的双键打开进行自
见表8—7。该体系采用石墨纤维增强的复合材料性能见表8—8。由基型固化反应,并形成网络结构,而且Miachacd加成反应后形成的线型聚合物中的仲氨基还
以与聚合物上其余的双键进行进一步的加成反应。此反应也称为扩链加成反应,该反应主要在预表8—7 DABPA改性BMI树脂固化物的性能
聚过程进行,扩链增大BMI树脂双键间的距离,减小了交联密度,从而提高了韧性,同时也改善体系1
体系2
体系3 性能 体系1
体系2
体系3 性质
弯曲强度/MPa
166 了树脂的溶解性和固化工艺性。
184
154 拉伸强度/MPa
4.0
3.98 (3)环氧树脂改性BMII 环氧树脂改性BMI是一种开发较早且比较成熟的一种方法。环氧树
25℃ 81.6 93.3 76.3 弯曲模量/MPa
3.95
压缩强度/MPa 205
209 脂主要用于改性BMI体系的工艺性和增强材料之间的界面黏结性,同时也明显改善了BMI树脂体
-
204℃ 39.8 71.3 一
压缩模量/MPa 2.38
2.47 系的韧性。环氧树脂本身很难与BMI单体反应,改善BMI体系韧性的途径主要有以下两种。
拉伸模量/GPa
25℃ 4.3
3.9 HDT/C
285
295 ①同时加入二元胺和环氧树脂。在这类体系中,BMI和环氧树脂通过与二元胺的加成反应而
11
204℃ 2
2.7 T,TMA/C
273
282 287
断裂伸长率/% 发生共聚,共聚反应的最终结果除形成交联网络外,BMI也被部分二元胺和环氧链节“扩链”,因
25℃ 2.3 3.0
2.3 而BMI体系的韧性得到了明显的提高。
204℃ 2.3 4.6
一 环氧树脂与BMI的反应在一般条件下不易进行,因此,环氧树脂、BMI通过与二元胺(DDM
表8-8 DABPA改性BMI树脂石墨纤维复合材料的性能
或DDS)的加成反应进行共聚。用这种方法得到的预聚物易溶于丙酮,预聚物具有良好的黏性和
性能 体系1
体系2 性能
体系1 体系2
成型工艺性。
层阿剪切强度/MPa弯曲强度/MPa
②合成具有环氧基团的BMI,这种方法属于内扩链。该方法是从BMI的分子结构出发,通过25℃
113 123
25℃ 1860
延长BMI分子中两个马来酰亚胺(MI)间R链的间距,并适当增大链的自旋和柔韧性,以达到降177℃
75.8 82
177℃ 1509
低固化物交联密度、减少链的刚性、改善树脂韧性的目的。反应方程式如下:232℃
59 78
177℃(湿)中 1120
|
177℃(湿)①
52 53
弯曲模量/GPa 25℃(老化)
一 105
25℃ 144
COOH- OH
177℃(老化) 55
177℃ COOH
144 177℃(湿)
142 ①71℃,95%湿度下放置2周。
②232℃老化1000h。
H H
除了DABPA改性BMI外,许多烯丙基化合物可用来改性BMI,例如二烯丙基双酚S、二烯
丙基双酚F、烯丙基芳烷基酚、烯丙基醚酮树脂、烯丙基酚环氧树脂、烯丙基线型酚醛树脂和N—OH
OH 烯丙基芳胺等。
为了降低吸湿性和提高韧性,在其改性树脂体系中加入了二氰酸酯化合物。该树脂体系突出
DABPA改性BMI树脂由于异丙基的存在而使其热氧稳定性较差,在高温下易分解而使其耐
的优点是具有类似于环氧树脂的优良工艺性,并在93~132℃之间具有良好的湿热性能。
热性降低。而采用二烯丙基双酚S,用它来改性BMI,改性后的树脂软化点较低,但溶解性较差,
BMI经环氧树脂改性后,其工艺性能和对增强材料的黏结性能有较大的提高,同时也增加了
力学性能同XU292体系基本接近,韧性一般,冲击强度和Gkc值分别为13.4kJ/㎡和160J/㎡,
BMI树脂的韧性。但是环氧树脂的加入往往会降低BMI树脂的耐热性。
它的耐热性很好,热变形温度(HDT)和Tg分别为295℃和309℃。
(4)热塑性树脂增韧改性BMI 用耐高温的热塑性树脂(TP)改性BMI树脂具有较好的增韧
芳烷基酚树脂具有优异的绝缘性能、耐热性能、力学性能和良好的工艺性,为此人们提出了
效果,同时不会降低其耐热性能和力学性能。影响增韧效果的主要因素有热塑性树脂的主链结构、
在芳烷基酚树脂上引入烯丙基基团,用于改性BMI树脂,烯丙基芳烷基酚改性的BMI树脂具有优
分子量、颗粒大小、端基结构、含量以及所用溶剂量的种类和成型工艺等。一般来说,用于BMI
异的综合力学性能和耐热性,冲击强度和G1c值分别为17.6kJ/㎡2和169]/㎡2,HDT和Tg分别
增韧的TP应满足以下要求。
为309℃和325℃.
①TP应具有良好的韧性。改性树脂的韧性与TP的韧性成正向关系,TP韧性越高,改性树为改善BMI树脂对纤维的浸润性和黏结能力,可采用含有较多—OH的烯丙基酚环氧树脂
脂的韧性也会相应提高,TP的韧性顺序:聚砜(PS)>聚醚砜(PES)>六氟聚酰亚胺(6FPD)。(AE)对BMI进行改性,克服了一般DABPA改性BMI体系制备的预浸料软而不黏的不足,另外。
②TP应具有良好的耐热性。当温度接近Tg时,改性树脂力学性能大幅度下降。一般选择具120
121
聚合物基复合材料
第8章 聚酰亚胺树脂
韧性的方法。
④TP要具有活性端基,活性端基能更好地发挥出TP对BMI增韧作用。具有的,分子是大,请有作用不明显,分,工批,一的的的
(5)氰酸酯树脂改性BMI 不论用二元胺扩链改性还是用烯丙基类化合物改性,其基本原理都是通过降低树脂交联密度来提高韧性,往往都是以不同程度地损失材料的刚性和耐热性为代价;
好的溶解性。
而采用氰酸酯(CE)改性BMI树脂体系可显示出不同的特点。氟酸酯树脂是一类带有—OCN官能团的树脂,其固化物具有良好的力学性能、耐热性能及优异的介电性能。20世纪80年代中期开始,氰酸酯树脂以其优异的综合性能受到人们的青睐,其性能介于环氧和BMI体系之间,兼有
。④的是,穿于N日交积体系中形成半互穿同络,随TP在体系中含
环氧树脂优异的工艺性能和BMI树脂的耐热性能,同时阻燃性能和介电性能优良,吸水率很低。氰酸酯改性BMI可在保持BMI体系具有良好的耐热性基础上,提高复合体系的韧性,同时改善体
用用的的形方物主要有度(PES、果据蔗亚胶(PE)、聚海因(PH)、改性黄组
系介电性能及降低吸水率等。其典型例子是BMI和氰酸酯树脂的共聚物(BT)树脂在高性能印刷
(PEK—C)、改性聚醚砜(PESC)和聚苯并咪唑(PBI)等。水C、或性聚是药塑性的芳杂环谐,具有化异的时高温和耐低温性能,工。为4300,
电路板中有着很好的应用。
氰酸酯改性BMI树脂的机理一般认为有两种:一种机理认为是BMI和氰酸酯共聚:另一种机理认为BMI与氰酸酯形成互穿网络而达到增韧改性效果。CE与BMI发生共聚的反应机理并未被实验所证实。
等,PBI改性BMI树脂的性能见表8—9。日本 Mitsubishi推出的BT树脂是通过烯烃取代CE改性BMI而取得的。通过烯烃取代的CE
表8—9 PBI改性BMI树脂的性能在催化剂存在下发生三聚反应,形成耐热性能、介电性能好的三嗪环,然后柔性的烯丙基或丙烯
2■ 基与BMI发生加成反应,形成的共聚树脂耐热性、柔韧性更好。将三嗪环和烯丙基同时引人双马
1廿
项目 材料及性能
33.35 来酰亚胺树脂中,基本上可以保持双马来酰亚胺树脂的耐热性,而且可改善其柔韧性和电气性能。
Marrimid 5292B/% 30
(6)橡胶改性双马来酰亚胺 在BMI树脂中添加少量带活性端基的橡胶,可以大大提高其抗66.65
60 冲击性能。橡胶在树脂固化过程中析出成为分散相,形成硬连续相—软分散体系,达到增韧的目
配方 Compimide 795/%
(质量分数) PH(粒径10μm)/%
10 的。然而,这种方法导致体系耐热性损失过大。与橡胶增韧环氧树脂相似,BMI树脂也可以采用
T,(DMTA,干态)/℃ 251
250 液体橡胶作为第二相增韧。报道用的液体橡胶有端羧基丁(CTBN)、端烯基丁腈(VTAB)、端氨
室温模量/GPa 4.53
3.97 基丁腈(ATBN)和有机硅烷,适当加人橡胶可改善BMI树脂的冲击强度、断裂性能和断裂伸
模量下降一半时的温度/℃211
211 长率。
T.(DMTA,湿态①)/℃ 182
8.3.1.5 双马来酰亚胺树脂的应用175
性能 3.86
3.6 双马来酰亚胺的耐热性优于环氧树脂,而经改性的BMI工艺性能可与环氧相当,特别是双马
室温模量/GPa 来酰亚胺材料的耐湿/热性能优异,这使航空航天部门给予极大的关注,可用BMI树脂作碳纤维
模量下降一半时的温度/℃151
150 复合材料的基体而代替或部分代替环氧树脂。将改性BMI材料用在航天飞机上,其重量比合金轻
Gx/(/m) 128
272 6.0%左右。
吸水率/% 3.24
3.93 (1)航天航空领域的应用 双马来酰亚胺具有阻燃、耐高温、低毒特性,经改性的BMI加工
①71℃,14天,100%湿度,性亦好。它们可做成蜂窝结构的平板材料,用于飞机地板、隔离墙、盥洗室材料、排气系统管子
表8—9所列数据表明,BMI经PBI树脂增韧后,断裂韧性Gic值有较大提高,而对Tg、模量等部件。与碳纤维复合,用于军用机或民用机或宇航器件承力或非承力结构件,如机翼蒙皮、尾
等影响不大。 翼、垂尾、飞机机身和骨架等。双马来酰亚胺在航空工业中的最初应用之一为Rolls—Royce公司制
哈尔滨理工大学陈宇飞等采用双烯丙基化合物(双酚A双烯丙基醚、二烯基双酚A)增韧双造RB—162发动机,其压缩机壳、转子翼片及定子翼轮都由玻璃纤维增强的双马来酰亚胺制造。这
马来酰亚胺树脂,该化合物提供了化学反应环境且一定程度上改善了材料的脆性;采用热塑性树些部件可以在240℃使用。美国空军战斗机F—22和RAH—66“卡曼奇”式武装直升机都大量使用
脂聚醚砜(PES)、聚醚醚酮(PEEK)为增韧剂使双马来酰亚胺的韧性提高得更显著;采用超临了双马来酰亚胺的复合材料,空重13.6t的F—22,热固性复合材料占结构重量的25%,几乎覆盖
界流体方法修饰无机纳米氧化物(二氧化硅、三氧化二铝)或有机化改性的蒙脱土(OMMT)并了飞机的全部外表面,其中大部分是双马来酰亚胺树脂复合材料。
将其掺杂于双马来酰亚胺树脂中,同时制备了三相复合材料。该复合材料既采用了“内增韧”法(2)电子电器方面的应用 BMI有显著的耐湿/耐热性能,尺寸稳定性高,热膨胀系数低,其
也利用了“外增韧法”,其微观形貌呈现多相结构,其中双烯丙基化合物是内增韧剂,与BMI核树脂有希望代替环氧树脂制造多层结构线路板,也可以作高温浸渍漆、层压板、模压塑料等,用
于电器的绝缘。 脂发生了化学反应形成了新的化学结构在复合材料中以基体相即连续相存在;而无机纳米相
(3)耐摩擦和磨损材料 用作金刚石砂轮、重负荷砂轮、刹车片等,也可用作热绝缘材料PES相和PEEK相在复合材料中是分散相即增强体,以“海岛”结构存在,是两种外增韧剂,且
(防热)、外层涂料等。与聚合物基体形成了一定的界面微区,界面作用的存在有利于复合材料的各项性能。该复合材料
在的热性能、力学性能及介电性能均有一定程度的提高,可作为高性能树脂基材料,并扩大其在8.3.1.6 双马来酰亚胺树脂的发展趋势及前景
航天、军工及民用方面的应用。该材料的研究是聚合物基复合材料中具有一定代表性的提高材目前,BMI材料的探索研究已取得很大进展。然而不仅在材料的结构与性能的关系上还有深
122 123
聚合物基复合材料
第8章 聚酰亚胺树脂相数的工作要做,而且在材料的开发及应用仍要进行许多探索,今后工作将主要集中在以下几
故称之为PMR15.反应物的物质的量比不同,所得到的预聚体的分子量也不同,由于Nadic酸
体很多,图8—3列出部分新型BMI的结构式。
后因化时产生挥发分,从而可制备密实的复合材料,但亚鞍化PMR应胶化反应完全,以的反应方程式如下:
PMR树脂
PMR—15树脂的合成是将BTDA和甲醇加热回流几小时,得到BTDE溶液,按配比将其他单加入BTDE溶液中即可得到PMR聚酰亚胺树脂溶液。如果BTDA和甲醇加热回流时间过长或BTDE溶液储存时间过长,会形成三甲酯或四甲酯,这将影响聚合过程中的链扩展从而使预聚体分子量降低。PMR—15的固化反应是按逆DielsAlder反应进行的,其固化反应式如下:
0
图8—3 部分新型BMI的结构式
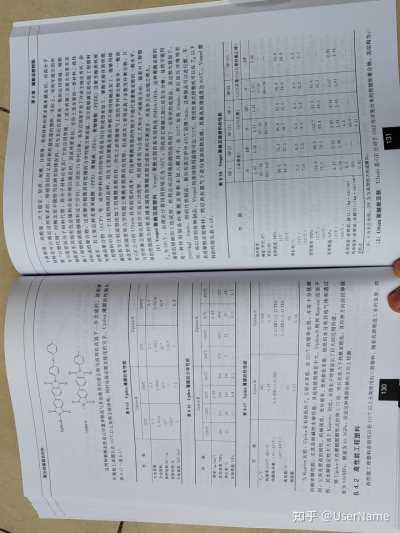
8.3.2 降冰片烯封端聚酰亚胺树脂PMR-15
降冰片烯封端聚酰亚胺树脂是指用二元胺、二元酸酐及封端单体合成的聚酰亚胺,其中主要8.3.2.2 PMR型聚酰亚胺的性能
的品种是美国NASA路易斯研究中心开发的PMR型树脂,PMR型树脂是芳香二胺、芳香四酸的(1)PMR聚酰亚胺树脂的物理性能 PMR方法可以通过调节预聚物的分子量,在一定程度
二烷基酯和纳迪克二酸的单烷基酯的甲醇或乙醇溶液。该溶液可直接用于浸渍增强材料,加热使上改变其玻璃化转变温度、力学性能、热氧化稳定性以及加工性能等。一般根据实际要求,在这
其发生亚胺化反应后制得预浸料,再经加热加压固化得到复合材料。PMR型聚酰亚胺树脂的特点些性能之间进行平衡从而选择适合的预聚物分子量。通常随着预聚物分子量的增加,材料的玻璃
是:使用低分子量、低黏度单体;使用低沸点溶剂;亚胺化反应在固化之前完成,固化时有极少化转变温度下降,层间断裂韧性增加,复合材料的热氧化稳定性提高。此外预聚物的分子量对其
的挥发物产生。用它可以制造出孔隙率小于1%的复合材料。熔体黏度影响很大,在一定程度上影响复合材料的加工温度、时间和压力等参数,并最终影响复
由于PMR树脂是在低级醇中形成溶液,是一种单体的溶液,所以可以做到高浓度、低黏度,
合材料的力学性能和热氧化稳定性。Leung研究表明,随着PMR—15预聚物分子量的增加,固化
可直接用来浸渍纤维和织物,并进行复合材料的成型固化,得到耐热和高力学性能的先进复合材
后聚合物的交联密度下降,但是由于封端剂含量下降,所以树脂的热氧化稳定性提高。表8—10为
料,聚合和交联反应在加工过程中进行。由于采用低分子量的单体进行反应,尤其在加工的初期
国内外一些商业化的PMR聚酰亚胺树脂的性能。
阶段,熔体黏度很低,和纤维表面能很好地结合,同时也可以在低压下加工。由于使用低沸点的溶剂,所以在加工过程中容易除去,降低了制品的空隙率。PMR—15聚酰亚胺树脂具有成型工艺
表8—10 国内外几种PMR聚酰亚胺树脂的性能
相对容易、热氧化稳定性好、力学性能优良和高温强度保持率高(288~316℃)等优点。但其也
树脂牌号
性能 典型值
实验方法及条件
存在明显的缺点:①制备厚、复杂构件时树脂流动性不足;②在成型时有可能会形成裂纹;
密度/(g/cm3)
1.36 GB/T 1636-2008
③原料(MDA)的毒性较大,影响操作人员的健康;④在317℃以上热氧化性能较差。特别是环
玻璃化转变温度/℃
GB/T 1040.1-2018 304~320
境问题的限制,使得PMR—15的应用受到限制,人们进行了一系列的工作,以寻求代替MDA制
KH304
拉伸强度/MPa 36
备PMR树脂的途径。3
拉伸模量/GPa 8.3.21 PMR型聚酰亚胺树脂的合成
起始分解温度/℃ 412
520 TGA
T■/℃ 芳香族聚酰亚胺的合成通常是通过芳香族二酸酐与二胺的缩聚反应来进行的,合成PMR聚
T.10/℃ 565
酰亚胺的单体通常有芳香二胺、芳香二酐和降冰片烯酸酐(又称Nadic酸酐,NA)等。可以通过
密度/(g/c㎡)
1.34
285~305 DSC
3种单体的不同物质的量比调节中间体酰胺酸和聚酰亚胺预聚体的分子量:纳迪克二酸单甲酯
玻璃化转变温度/℃
45 GB/T 2568-1995
(NE)、4.4'—二氨基二苯基甲烷(MDA)和3,3',4,4'—二苯甲酮四羧酸二甲酯(BTDE)。反应物之
LP-15
拉伸强度/MPa 3.2
间的物质的量之比为NE:MDA:BTDE=2.000:3.087:2.087时,得到的预聚体分子量为1500,
拉伸模量/MPa
TGA 415
124 起始分解温度/℃
125
第8章
聚酰亚胺树脂 聚合物基复合材料
续表 表8—11 PMR—15复合材料的性能
典型值 实验方法及条件
何脂牌号 性别
284 增强纤维
纤维体积
玻璃化温 弯曲强度/MPa
玻璃化转变温度/℃ 分数/%
度Ta/℃ 弯曲模量/GPa
层间剪切强度/MPa
起始分解温度/℃ >400
TGA 室温
446 316℃
室温 316℃
PMR-15 T3/C
Celion6000 60
338 室温
316℃ 1846
1096
T3/C 87
Celion6000(无浆料) 58
340
1758 91
110 55
115
断裂性/(J/㎡) 862
114
密度/(g/c㎡) 1.35
Celion6000(环氧浆料) 59
330
1724 116
53
玻璃化转变温度/℃ 265
800
LARC RP46 G40—700(无浆料)
57.5 340
1510 119
45
断裂韧性/(J/㎡)202
814
G40—700(环氧浆料)
59.8 335
1379 95
48

















765
降冰片烯酸酐封端的聚酰亚胺的结构与性能有很大的关系。LaRC—13是以m,m'—MDA,T40R(无浆料)
62 340
1138
807 90
44
纳迪克酸酐、对苯甲酮四甲酸酐(BTDA)为原料,合成酰胺酸预聚物,分子量1300,在IM6(无浆料)
57.5
335 61
















43 1772
786
120~200℃下亚胺化,在280~300℃有压力的情况下固化,其熔程为200~240℃,熔化后
Celion600(环氧浆料) 53
106
43
1634 1260 100 88
黏度低。 Celion600(AvimidN浆料)
61 333
92
43
1670
(2)PMR聚酰亚胺树脂的流变性能 图8—4为PMR聚酰亚胺树脂的熔体流变曲线,显示97
了PMR—15聚酰亚胺树脂的凝胶特性。从中可见,LaRC—RP46比PMR—15的凝胶温度高。420
0-8800 (3)PMR聚酰亚胺树脂的耐热性能 聚合物在高温下会发生交联、氧化降解等化学反应,
使其性能变坏,同时也会使物理状态发生变化,如密度增加、脆性增加。因此,聚合物的热氧-20-
400
化稳定性是衡量其耐热性大小的重要性能。380
40
图8—5是PMR—15聚酰亚胺复合材料在不同温度下热老化后的质量损失曲线。由图看出,在0-316℃
-60 -0-343℃
360 316℃下热老化1000h后,PMR—15聚酰亚胺复合材料的质量损失仍小于10%(质量分数),但
-371℃
在371℃下热老化200h后,其质量损失已达20%(质量分数)。因此,PMR—15聚酰亚胺复合材-80
340
-100 320
料的长期使用温度应低于316℃。0
200 400 600 800
1000 0
200 400 600
10 时间/h
800
LaRC-RP46 20
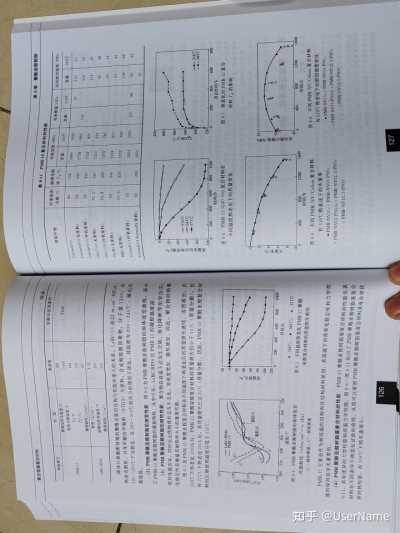
图8-6 PMR—15/G30—500 复合材料在
老化时间/h
PMR-15 图8-7
热老化对PMR—15复合
0
10% 不同温度热老化下的质量变化
材料Tg的影响
-20
10% -40
损
凝胶点 -60
凝胶点 -80
额
10 55
-100 -120
10 200 220
240 0
200 400 600 800 1000
義
260 280 300
320 时间/h
6
8
8 温度/℃
·316℃:343℃; 371'℃ 图8—4 PMR聚酰亚胺树脂的熔体流变
图8—5 不同温度热老化PMR—15聚酰10
45
性能曲线(1dyn/c㎡2=0.1Pa) 0
400 800
1200 1600
0 400
800 1200
1600
亚胺复合材料的质量损失曲线时间/h
时间/h
G'一储存模量;G”—损耗模量图8-8
不同PMR-NV/Celion复合材料 图8-9 不同PMR-NV/Celion复合材料
在316℃热老化下的失重率在316℃热老化下的剪切强度变化
PMR—15主要是作为耐高温的结构和次结构材料使用,其高温下的热氧化稳定性和力学性
·PMR-NV15:·PMR-NV15-PN5;
·PMR-NV15;o PMR-NV15-PN5; 能的保持是至关重要的。
PMR-NV15-PN10;·PMR-NV12.5-PN5; PMR-NV15-PN10;°PMR-NV12.5-PN5;
(4)PMR聚酰亚胺树脂基复合材料的性能 PMR—15聚酰亚胺树脂基复合材料的性能见表
PMR-NV12.5-PN10
PMR-NV12.5-PN10
8—11,具有优异的力学性能和高温力学性能。图8—6~图8—11给出了PMR聚酰亚胺树脂基复合材料在不同条件下热老化试验的曲线。从图可以看到PMR聚酰亚胺树脂基复合材料具有非常优异的热性能,在316℃下热失重很小。
126
127 T(DSC)/C
层间剪切强度/MPa质量变化(初始质量)/%
失重率/% G:G"(dyn/c㎡)
聚合物基复合材料
第8章 聚酰亚胺树脂
Kapton 薄膜的结构如下:
120
0011
011
E
1000 力学性能、电性能及热性能见表8—12~表8—14所列。此外Kapion薄膜还具有优异的耐辐
K 8
100 射,耐溶剂等性能,涂有含氟聚合物的Kapton具有黏结性和密封性,情膜还具有优异的耐物
900% Kapion品种。填充氧化铝的Kapton是具有高导热性的绝缘薄膜。
1600 90 0 400 800 1200 1600
400 800
1200 时间/h
表8—12 Kapton 薄膜的力学性能和电性能
0000
时间/h
图8-10 不同PMR-NV/Celion 复合材料 316℃热老化下的弯曲模量变化
性
能 -195℃
25℃ 图8-11 不同PMR-NV/Celion复合材料在
200℃
在316℃热老化下的弯曲强度变化
·PMR-NV15PMR-NV15-PNS:PMR-NV15-PN10;
·PMR-NV15:·PMR-NV15-PN5:&PMR-NV15-PN10
相对密度 1.42
PMR-NV12.5-PN51PMR-NV12.5-PN10 抗拉强度/MPa
246.5 176.0
119.7
。PMR-NV12.5-PN5:PMR-NV12.5-PN10 伸长率/%
2 70
90
抗张模量/GPa 3.59
3.03 1.83
8.3.23 PMR聚酰亚胺树脂的应用510
初始抗撕强度/(g/mil)
介电强度/(kV/mil) 10.8
7 5.6
介电常数 3.5
3.0
介电损耗 246.5
0.003 0.002
拓展阅读 体积电阻(50%RH)/0·cm
2 101
10
表面电阻/Ω 3.59
101
表8-13 Kapton 薄膜的热性能
8.3.3 乙炔封端聚酰亚胺
含炔基树脂近年来已成为热固性树脂研究的热点。此类树脂具有高反应性,在一定条件下,无
使用寿命 250℃/8a,275℃/1a.
熔点/℃ 如热、辐射等条件能加成聚合形成体型结构,固化过程中没有挥发性副产物产生,固化产物具
零强温度(1.4kg/15s)/℃815
300℃/100d,400℃/12h
有无气隙、耐湿热、热稳定性和性能保持率高的特点,从而为获得高热性能复合材料提供了可T/C
385 收缩率(250℃.30min)/%
0.3%
能性,目前,已研究和开发了一些新型的含炔基树脂如聚芳基乙炔树脂、含硅芳炔树脂、炔基热影胀系数
2.0x10-5 氧指数/%
37
聚酰亚胺、乙炔基封端的聚苯基喹嘎啉、聚芳砜、聚醚、聚苯井哪唑、聚苯并咪唑、聚苯并咪唑表8—14 各种Kapton薄膜的性能
喹啉等树脂,并对其结构和性能进行了表征。以下就新型的含炔基树脂做以介绍。Kapton 薄膜的型号
聚芳基乙炔树脂(PAA)是指二乙炔基苯经预聚而成的树脂。其主要特点是:①预聚物呈性能(23℃)
XT(氧化铝) XC-10
200X-M25 100C09
液态或易溶、易熔的固态,便于复合材料成型加工;②聚合过程是一种加聚反应,固化时无挥200H
MD/TD (导电碳)
(滑石粉) (活性炭)
发物和低分子量副产物逸出;③树脂固化后通常呈高度交联结构,耐高温性能优异;④分子结抗拉强度/MPa
239.4 140.8/119.7
112.6 140.8
140.8
构仅含C和H两种元素,碳含量达90%以上,热解成碳率极高。伸长率/%
90 30/30
40 35
59
介电强度/(kV/mil) 6.0
4.0 3.4
0.8
8.4 聚酰亚胺薄膜、塑料及纤维介电常数
3.4 3.4
11 3.9
介电损耗 0.0025
0.0024 0.081
0.012 一
表面电阻/0 1017
1010 -
10
8.4.1
薄膜 体积电阻/0·cm
101 10
1012 10■
10% 一
-
热导率/[W/(m·K)]0.155
0.24
吸湿率(100%RH)/%3
5 3.7
一
0.5
(1)Kapton 薄膜 薄膜是聚酰亚胺作为材料最早开发的产品之一,是杜邦公司在20世纪高温收缩率(400℃)/%
1 1.2/0.6
60年代初发展起来的Kapton薄膜。除了在俄亥俄州外还在日本建厂生产,总产量在3000~注:1mil=0.0254mm,下同。
5000t。这是由均苯四酸二酐和4.4'—二苯醚二胺在极性溶剂如DMF、DMAc、NMP等溶剂中缩日本Unitika 公司的Echigo用四氢呋喃/甲醇为溶剂合成PMDA/ODA聚酰胺酸溶液,并由
聚,然后将得到的聚酰胺酸溶液在基板上涂膜,干燥后再在300℃以上处理完成酰亚胺化。根据此获得厚度为300~500μm的透明薄膜。由非质子极性溶剂难以得到厚度为200μm以上的透明
化学酰亚胺化方法,1968年杜邦发展了凝胶成膜法,将其用于单向和双向拉伸的薄膜上,即在薄膜,因为高沸点的溶剂难以除去,会使薄膜变得不透明。
冷却的聚酰胺酸DMAc溶液中加入乙酐和β—甲基吡啶,然后在热鼓上形成含有大量溶剂,大部(2)Upilex薄膜 Upilex薄膜是日本宇部公司在20世纪80年代发展起来的聚酰亚胺薄膜,
分己酰亚胺化了的凝胶的膜。在室温拉伸1倍,然后在张力下加热(最高温度为300℃)去除溶其结构有两种:Upilex-R及Upilex-S,其结构如下:
剂得到薄膜。这种方法也同样适用于其他类型的聚酰亚胺薄膜。
128 129
弯曲模量/GPa
弯曲强度/
第8章
聚酰亚胺树脂
度,高模量,尺寸稳定、轻质、耐服、自利滑、密封材料的要求越来意迫切,而离分子聚合物基复合材料
材甲“以如代钢”的概念是不能恰当反映实际情况的,因为无论在资源变际上,早些年提出的所形铁甚至其他有色金属和合金所难以满足的,因此所要求的,实际上用机被工业提出的要求是
Upilex-S 诉组成的聚合物,这类聚合物既具有优越的力学性能又耐高温,完全能够清多主链由方环,条
这两种聚酰亚胺是以对氯苯酚或与其他酚类的混合物为溶剂在高温下一步合成的。该溶液
在基板上成膜后在300℃以上加热去除溶剂,同时也保证酰亚胺化的完全。Upilex薄膜的性能见维液品聚合物(LCP)等,这些材料的共同特性是容易用注射成型来加工,聚低类热级
工程塑料中是一个比较特殊的品种,因为主要的聚酰亚胺品种都不能用熔融法加工,在够俗
融法甚至注射成型法加工的聚酰亚胺在性、价比上必须能够与上述的芳环聚合物竞争。一般说表8—15~表8—17。
来在使用温度和力学性能上聚酰亚胺都具有优势,但是成本上要明显高于多数芳环聚合物,只表8—15 Upilex薄膜的电性能
有GE公司的Ultem具有较低的成本,但这种聚酰亚胺的性能并不能代表聚酰亚胺的一般水平,Upilex R
Upilex S
能 25℃
与芳环聚合物比较并不显示出优势。然而成本是与合成工艺和生产规模有关的,随着对工程塑性
25℃ 200℃
200℃
介电常数 料的性能提出的要求越来越高和聚酰亚胺合成技术的不断进步,其竞争力也会随之增大。
3.5 3.2
3.5 3.3
介电损耗 0.0014
0.0040 0.0078
(1)Vespel 聚酰亚胺塑料 Vespel聚酰亚胺的基本结构是PMDA/ODA,这种聚酰亚胺的0.0013
体积电阻/Q·cm 107
10■ 1017
1015 T.为385℃,由理论计算得到的熔点为592℃,因此在它熔融之前已经发生分解,显然不能用
表面电阻/0 >10
>1016
介电强度/(kV/mil) 7
7.1 7
通常的熔融加工法成型。杜邦公司以特殊的烧结法得到了一定形状的成型品。其过程大致如下:6.8
将一种特别制备的聚酰亚胺粉末加入模具中,在300℃加热10min,然后加压并维持在
表8—16 Upilex薄膜的力学性能20000kgf、2min,得到片状制品,最后在真空炉中450℃处理5min。这种制品可以进行磨、车、
Upilex-R Upilex-S
切,钻以得到各种制品。Vespel的连续使用温度可达315℃。使用柱塞式挤塑机可以在Tg以下
-269℃ -196℃ 25℃
300℃ -196℃
25℃
300℃ 获得聚酰亚胺棒材,然后再在氮气下进行复杂的热处理,其最高处理温度达400℃。Vespel塑
性
能 -269℃
密度/(g/c㎡) 一
1.39 -
1.47 料的性能见表8—18。
一
一
抗拉强度/MPa 300
270 250 200 570 500 400 220
伸长率/% 15
40
130 表8—18 Vespel 聚酰亚胺塑料的性能
190 7 11 30 48
抗张模量/GPa 3.8
2.1 9.0
3.5 SP-1
SP-21 SP-211 SP-1
SP-21 SP-211
-
一
一
表8-17 性
能 (未填充)
(15%石墨) (15%石墨+10%聚四氟乙烯)
Upilex 薄膜的热性能S
DF S DF
S DF
Upilex R Upilex-S
1.43 1.36 1.51 1.43
1.55 1.46
性
T./℃ 陈
相对密度
收缩率(250℃.2h)/%285
>500 硬度(罗氏,E)
45~58 32~44
5~25
一
-
0.18 0.07
罗氏(M) 92~102
- 82~94
69~79
线膨胀系数 (20~250℃)
2.8(MD),3.2(TD) 1.2(MD),1.2(TD)
拉伸强度/MPa (20~400℃)
23℃ 88.0
73.9 66.9 63.4
45.8 52.8
氧指数/% 55
1.5(MD),1.5(TD) 37.3
38.7 31.0
24.6 24.6
烟指数 66
260℃ 42.3
0.07 0.04
仲长率/% 4.5
6.0
3.5 5.5
(23℃) 7.5
8.0 7.0 7.0 2.5 5.2
3.0 5.3
(260℃) 与Kapton比较,Upilex—R有较低的Tg且吸水率低,在250℃收缩率也低,并有十分优越
抗弯强度/MPa 的耐水解性能,尤其是耐碱性水解性能。其他性能则相差不大。Upilex—S则和Kapton完全不
98.6 112.7 91.5
70.4
(23℃) 133.8
63.4 49.3
35.2 -
(260℃) 77.5
58.5 同,它具有更高的刚性、机械强度,低收缩率、低热膨胀系数、较低的水分和其他气体和透过
性。其水解稳定性大大高于Kapton,因此,在微电子领域显示了巨大的应用价值。抗奇模量/GPa
2.53 3.87 3.24
3.17 2.82
(23℃) 3.17
2.61 1.83
1.41 1.41
将Upilex—S在聚酰胺酸形式拉伸1.75倍,然后在张力下热酰亚胺化,其拉伸方向的拉伸强(260℃)
0.76
1.48
度为986MPa,模量为49.3GPa,但是这种薄膜在横向方向上较脆。8.2
-
4.4
(日/月·3)/(口指。躲事出)经事是据
8.4.2 高性能工程塑料抗冲强度(悬臂梁,无缺口)/(kg·cm/cm)
163 43.6
泊松比 0.41
0.41
注:S为无方向性;DF为与成型的方向成横向。
高性能工程塑料是指可以在150℃以上长期使用的工程塑料。随着机器制造工业的发展,对
(2) Ultem 聚醚酰亚胺 Ultem是GE公司于1982年开发出来的热塑性聚合物,其结构为:
130 131
聚合物基复合材料
第8章 聚酰亚肢树脂
聚酰亚肢纤维与开美拉纤维比较有更高的热稳定性、更高的弹性模量、低的吸水性,可望CH
在成积物或做成无纺布,用在高温、放射性或有机气体或液体的递保立胶纤维可编或级
装等, 的于Uhem中含有双醇A残基,耐溶剂性较差,T。仅为217℃,因此虽然具有很好的加正
聚酰亚胺纤维至今仍未有较大规模的生产,其原因是:第一,在目前的技术水平,芳香聚酸酸纤维(杜邦公司)已基本满足要求,对于性能更离的纤维,并非许多技术水平,方容限
按在排气挤出机中进行无溶剂聚合,或由双(4硝基酰酰)二亚胺和双酚A直接缩聚而榫。二,聚酰亚胺纤维的成本太高是阻碍发展的主要原因。
表8—21 由均苯二酐和对苯二胺及3,4—二苯酸二胺所得到的纤维
其性能见表8—19。组成
牵伸比 牵仲温度/℃
强度/(g/d)
仲度/% 模量/(g/d)
表8-19 Ultem的性能 未牵仲
1.8 12.5
20
型
号 4.0
550 12.6
7.1 354
条件 4.75
575 14.7
4.7 427
性别 单位
1000 2100
2200 2300
4.7
600 14.5
1.27 1.34
1.42 1.51
PPD/3.4'-ODA(25/75) 6.0
3.7 492
相对密度 23℃.24h
0.25 0.28
0.26 650
12.8 2.8
519
吸水率 %
0.18 6.1
675 15.6
3.3 570
23℃,浸渍饱和
1.25 1.0
1.0 0.9
6.8
186kg/cm 200
207 209
210 700
13.1 3.0
592
热变形温度 23℃
6.2x10- 3.2x10-
2.5x10- 10.0
700 15.5
3.4 534
2
线膨胀系数 /℃
2.0x10-1
拉伸强度 MPa
23℃ 107
122 143
163 5.0
750 3.6
3.7 168
伸长半 23℃
60 6
3 3
3.6
500 9.5
8.6 248
%
拉仲模量 MPa
23℃ 3060
4590 7040
9180 4.5
550 8.9
2.7 404
抗弯强度 MPa
23℃ 148
205 214
235 3.4'-ODA
4.0 550
10.7 5.5
302
抗弯模量 MPa
23℃ 3370
4590 6330
8470 4.0
575 8.3
4.2 321
抗压强度 MPa
23℃ 143
163 163
抗压模量 MPa
23℃ 2960
3160 570
3880
悬臂梁抗冲强度 kg·cm/cm
23℃,无缺口
370
49 49
44 因此,随着聚酰亚胺本身技术的发展,尤其是合成技术的发展和在其他领域应用的扩大,
9 10
聚酰亚胺的成本会有大幅度的降低。同时各个技术部门本身的发展也将会对于更高性能的纤维
9
23℃,无缺口 130
注:Utem1000为未填充;Ultem2100为10%玻璃纤维;Ultem2200为20%玻璃纤维;Ultem2300为30%玻璃纤维。的需要迫切起来。可以认为聚酰亚胺纤维的研究是超前的工作,聚酰亚胺纤维仍然是未来的
8.4.3 聚酰亚胺纤维材料。
聚酰亚胺纤维的研究开始于20世纪60年代美国和苏联。我国从事聚酰亚胺纤维的研究在8.5 聚酰亚胺胶黏剂
60年代中期,由华东化工学院和上海合成纤维研究所合作由均苯二酐和二苯醚二胺的聚酰胺酸
干纺而得。第1个聚酰亚胺纤维的专利是由杜邦的Irwin在1968年发表的,是由均苯二酐和聚酰亚胺作为胶黏剂的胶黏对象主要有3类:金属(钛、铜、铝及钢等)、非金属(硅片、
ODA及4,4'—二氨基二苯硫醚在DMAc中得到聚酰胺酸,干纺成聚酰胺酸纤维再在一定的张力玻璃及磨料如金刚砂、氮化硅等)及聚合物(如聚酰亚胺自身)。要达到良好的黏合,除了选择
下转化为聚酰亚胺,最后再在550℃牵伸得到聚酰亚胺纤维。PMDA—ODA也可以湿纺在吡啶溶合适的胶黏剂之外,基底的表面处理也很关键。聚酰亚胺结构中带有多个羰基,并且具有强的
液中成纤,然后热处理转化为聚酰亚胺。取向的纤维在400℃空气中经2~3h其强度仍保持传荷作用,这些因素都是作为胶黏剂的良好条件。
30%~40%.PMDA—ODA纤维的性能见表8—20。聚酰亚胺对聚合物的黏合主要针对的是与聚酰亚胺自身的黏合及与环氧类聚合物的黏合,
由均苯二酐(PMDA)和二苯醚二胺(ODA)得到的纤维尤其对于印刷线路板的制造,目前主要还是利用聚酰亚胺薄膜通过一层胶黏剂与铜箔的黏合而
表8-20
转化方法 牵伸比
牵伸温度/℃ 制得。这就需要聚酰亚胺薄膜与胶黏剂之间有很好的黏结力,其间的剥离强度要求达到10N/cm
组成
PMDA/ODA
热 强度/(g/d)
伸度/% 模量/(g/d)
1.5x,1.5x 左右。所用的胶黏剂有环氧类或丙烯酸类聚合物,但由于这些胶黏剂的耐热性不够高,通常难
PMDA/ODA 热/吡啶
1.5x 550
3.5 11.7
50
PMDA/ODA 热 1.5x,2.25x
600 24.0
以在70℃使用,所以对更耐热的胶黏剂,尤其是聚酰亚胺类胶黏剂的要求越来越迫切。
5.3
PMDA/ODA
热 575
6.6 49
1.5x,2.05x
525 77
聚合物对聚合物的黏合是以大分子通过界面扩散后发生的缠结而实现的。聚酰亚胺对聚酰
9.0
1.6x 420
2.8 亚胺的黏合决定因素有:聚酰胺酸通过界面的扩散;两层薄膜的固化机制;酰亚胺化后界面处
PMDA/SDA
热 5.1
9.0 68.5
注:SDA为4,4—二氨基二苯硫醚,25.7
31 的分子结构及聚集态。
当聚酰胺酸溶液涂到聚酰亚胺基底上时,后者可能会由于NMP的作用而发生溶胀,使前者
Irwin 还报道了由均苯二酐和对苯二胺及3,4'—ODA的共聚物在DMAc—吡啶中湿纺,部分热
能够穿透基底,在随后固化时发生化学结合(也有缠结),基底的堆砌密度越低,透入的可能性
酰亚胺化后,最后在高温下牵伸所得到的纤维,见表8—21。
132 就越大。对于PMDA(均苯四甲酸二酐)、BPDA(连苯二酐)、BTDA(酮酐),以PMDA得到
133
聚合物基复合材料
第8章 聚酰亚胺树脂
的果供要股的体因售位最大,BDA的维和度最小,三酸也同部可能的采合物的的
表8—24 扩散距离与固化温度的关系单位:μm
第二层固化温度/℃ 第一层固化温度/℃
150 200
300 的度取的,再及的养的能系数表则是小,结合力落大。当膨胀系数的差别较大
400
150 168
42 28
22
到高的黏结力。 的熟括方面按薄须是完全酰亚按化了的部质,尤其是Kaon或Upilex薄膜都具有很高的则
200 150
43 29
31
300 154
46 35
31
性,这种需反应,因此需要预先对聚酰亚脓薄膜的表而进行处理。处理方法有两种,一种400
156 47
37 30
Miwa等研究了各种聚酰亚胺对聚酰亚胺的黏合性能,在用水蒸气处理前后的黏合性能见表
KOH溶液在22℃处理1~90min得到聚酰胺酸钾,将多余的KOH用水洗去后,再用乙酸进行在准可以与胶黏剂作用的活性基团,以增加黏合能力。例如Lee等将Kapton薄膜用1mol
8—25.在完全酰亚胺化的聚酰亚胺上的黏合效果较差,而且受水蒸气的影响也较大,当第一层
质子化,在表面处理过的薄膜上旋涂上聚酰胺酸,干燥、固化,所得到的剥离强度见表8—22。聚合物仅以部分酰亚胺化则能够明显提高与聚酰亚胺的黏结强度,同时耐水蒸气的性能也大大
提高。第一层聚合物的固化温度与酰亚胺化程度及剥离强度的关系见表8—26,可见要得到高的
表8—22 聚酸亚胺薄膜90℃的剥商强度剥离强度,第一层聚合物的固化温度不能超过200℃,这时酰亚胺化程度达到50%左右,
KOH 处理时间/min 0
1 5
10 表8-25
剥离强度 单位:N/cm
剥离强度/(N/cm)0.3
4.0 8.5
12.6 在BPDA/ODA上,120℃
在半固化(200℃/h)的BPDA/PPD 在半固化(200℃/1h)的
水蒸气,100h 上,120℃水蒸气,100h
BPDA/ODA上,120℃水蒸气,100h
用碱处理薄膜所达到的深度越大,剥离强度越高,Buchwalter等用0.25mol/L的NaOH对聚合物
处理前
处理后 聚合物
处理前 处理后 聚合物 处理前 处理后
经过400℃固化的PMDA/ODA表面进行处理,其水解程度见表8—23。PMDA/ODA
4.41 2.45 BPDA/PPD
7.84 7.35 BPDA/PPD
7.64 6.86
BPDA/ODA 7.35 4.90 PMDA/ODA
7.35 7.64 PMDA/ODA
7.45 8.04
表8—23 PMDA/ODA聚酰亚胺的水解程度PMDA/BAPP
6.86 7.35 BPDA/ODA
7.35 7.45 BPDA/ODA
7.64 8.04
水解时间/h 水解分数/%
标称水解深度/nm BPDA/BAPP
4.41 2.45 PMDA/BAPP
7.35 7.25 PMDA/BAPP
7.45 7.84
BPDA/BAPP 7.25 7.35 BPDA/BAPP
7.64 7.55
0 0
0
14.6 16.0
表8-26 第一层聚酰亚胺固化温度与剥离强度的关系
1
3.5 38.1
41.9 固化温度/℃
酰亚胺化程度/% 剥离强度/(N/cm)
2 26.7
29.4
150 10
7.35
Yun等用胺的溶液处理聚酰亚胺薄膜表面以增加对环氧树脂的黏结力,胺的浓度一般为200
50 7.35
250 85
0.20
0.5%(质量分数),时间为1min。可用的胺有肼、乙二胺、1.3—丙二胺、已二胺等,干燥温度0.29
以100℃为宜。 300
90
100 0.20
350
另一种方法是用等离子处理,例如用N2/H2和NH3等离子处理,由于在处理后薄膜的表
面产生了可以与聚酰胺酸作用的氨基,从而使黏结强度增加。水蒸气等离子处理使PMDA/Ree等研究了BPDA/ODA与各种聚酰亚胺的黏合,BPDA/ODA的聚酰胺酸在80℃干燥
ODA聚酰亚胺表面的氧的浓度明显增加,生成了酮、羧酸及羟基等含氧的基团。未经等离子处后,氮气流中在150℃、200℃、300℃各处理30min,400℃处理1h。基底在旋涂前用等离子
理的薄膜对薄膜的90°剥离强度为0.49N/cm;处理后达到不小于8.82N/cm。另一种水蒸气等(300W/5min氧流速度:535cm3/min)处理。对于某些基底,需要使用氨丙基三乙氧基硅烷
离子处理的薄膜对薄膜的剥离强度为12.25N/cm,超过了18μm厚的薄膜的强度,在85℃、[y—APS,0.1%(体积分数)的乙醇/水(95/5)溶液]作为黏结促进剂。使y—APS的溶液以
81%相对湿度环境中放置1000h,剥离强度仍保留8.77N/cm。等离子处理也增加了薄膜的粗糙2000r/20s旋涂到基底上,然后在120℃热板上加热20min,见表8—27。
度,即增加了薄膜的表面积和反应基团,这些都有利于第二层聚合物的黏结。单位:N/cm
表8—27 对BPDA/ODA聚酰亚胺的剥离强度
如果采用的不是现成的商品薄膜,则可以用控制第一层聚酰亚胺固化程度来增加聚酰亚胺剥离速度/(mm/min)
样品结构(由上面下)0.05
0.1 0.2 0.5 1.0 2.0
5.0 之间的黏结力,即将第一层聚酰胺酸在较低温度下酰亚胺化使其达到不会因第二层聚酰胺酸的
涂覆而溶解,又能留下足够的酰胺酸部分可以与第二层的聚酰胺酸作用,以达到既可以相互穿
10.60
10.98 11.18 11.48
11.87 一
-
BPDA-ODA//BPDA-ODA 8.14
8.73 8.93 9.12 9.22 9.42
9.71
透,又可以使聚酰胺酸间发生交换,从而达到高的黏结强度。Brown等研究了第一层聚酰亚胺
BPDA-ODA//BPDA-ODA/PMDA-ODA
不能剥离
不能剥离
的固化温度对聚酰胺酸扩散深度的影响,由表8—24数据可见,对于PMDA/ODA的自黏,第一
不能剥离不能剥离不能剥离不能剥离不能剥离
一 BPDA-ODA/等离子处理/BPDA-ODA
11.38
12.36
层聚酰胺酸的固化温度以低于200℃为宜,同时第二层涂覆后采用高的固化温度对黏结也有利。
BPDA-ODA/等离子处理/底胶/BPDA-ODA
10.69 0.79
聚酰亚胺还经常与环氧树脂共用,以综合环氧树脂的易加工、高黏结性和聚酰亚胺高耐热
134 135
聚合物基复合材料
性的效果,将像的证分子聚低亚度作为环复树脂的圆化剂使用,另一类是在能亚胶结构上有效第9章 聚氨酯树脂
环氧基团。 据国等向PMIDA—ODA在THF/MeOH(80/20)10%的溶液中加入环氧材脂,涂膜后干燥,
可将面钢数的制的粘合效果见表828,可见适量的聚酰胶酸可以很好地促进环氧树脂的黏结性聚氨酯是聚氨基甲酸酯的简称,是主链上含有一NH—CO—基团的一类聚合物的通称。工
能。但在这种情况下,PMDA—ODA聚酰胺酸的酰亚胺化很可能是不完全的。业上,它是由多元异氧酸酯与多经基化合物通过聚合反应生成的。聚酯美软合物的通称。
表8—28 聚酰胺酸作为环氧树脂的固化剂维体、涂料、胶粘剂、纤维、合成皮革以及辅面材料等,它广泛应用于机电,有泡床塑料、
环氧树脂的固化剂 黏合强度/(N/cm)
环氧树脂的固化剂 黏合强度/(N/cm)
PMDA <20
80%聚酰胺酸 约5.0
ODA 约10
10%聚酰胺酸 100~200
展聚氨酯树脂工业。
陈宇飞等以均酐型酸酐、醚胺为反应单体,以纳迪克酸酐、马来酰酐、甲基纳迪克酸酐为9.1 原料
封端剂制备了热固性聚酰亚胺,并采用纳米氧化铝为改性材料制备了复合材料,分析了材料的
微观结构及力学性能、耐热性能和介电性能。制造聚氨酯的主要原料是多元异氰酸酯和含活泼氢的聚醚与聚酯多元醇。除上述两种原材
料外,聚氨酯树脂产品广泛地采用催化剂、交联剂、扩链剂、表面活性剂、发泡剂等助剂。这些助剂可以改进聚氨酯树脂生产工艺、降低成本、延长使用寿命、增加品种等。
9.1.1 多元异氰酸酯
低分子二异氰酸酯、三异氰酸酯是由脂肪族、芳香族二元胺、三元胺与光气在20~150℃下
反应制得: H2NRNH2+2COCl2→OCNRNCO+4HCI
常用的二异氰酸酯主要有六亚甲基二异氰酸酯(HDI)、2.4—甲苯二异氰酸酯、2,6—甲苯二
异氰酸酯(TDI)和二苯基甲烷二异氰酸酯(MDD).
CH1 CH
NCO OCN
-NCO
NCO
2.4—甲苯二异氰酸酯2.6—甲苯二异氰酸酯
OCN NCO
二苯基甲烷二异氰酸酶
甲苯二异氰酸酯中,2,4—异构体的活性较大。工业产品中,有单纯的2,4—异构体(例如
TDI—100),还有两种异构体的混合物,包括2,4—异构体与2,6—异构体之比为80/20的TDI—80及异构体之比为65/35的TDI—65,其中以TDI—80应用最普遍。几种二异氰酸酯的物理性质
见表9-1。
表9—1 几种二异氰酸酯的物理性质
性质 2.4—甲苯二异氰酸酯
六亚甲基二异氰酸酯 二苯甲烷二异氰酸酯
密度/(kg/㎡) 1217.8
1046 1185
(熔点) 21.8
-67 40
温度/℃
(沸点) 121(1.3kPa)
127(1.3kPa) 180~182(400kPa)
137
聚合物基复合材料
第9章 聚氨稲树脂
(1)聚醒 聚壁的端基为羟基。它是由环氧化烯经经开环聚合而得。环氧化烯经主要有环续表
2.4—甲苯二异氰酸酯六亚甲基二异氰酸酯
二苯甲烷二异氰酸酯
性质
140 它有多元醇与多元胶两种,起始剂中的活性氢数决定了聚的含能引起开环聚合的起始剂。
一
一
(四点) 402
一
(自燃湿度) 分子量。
-
多元醇起始剂有乙二醇、丙二醇、甘油、三羟甲基丙烷、季戊四醇、木糖醇等,如用环氧生产场所空气中的医限允许
0.5 0.05
0.5
浓度/(kg/m) 丙烷与乙二醇反应所得的聚醚产物为:
三异氰酸酯有三苯基甲烷三异氰酸酯(TTD:CH:OH
CH CH1
+(m+m)CH:-CH→CH2O(CH;CHO),H
NCO
CH:OH
OCN CH:O(CH:CHO).H
环氧丙烷与甘油的反应产物:CH1
NCO CH2O(CH:CHO),,H
NCO
NCO NCO
CH1 还有多苯亚甲基多异氰酸酯聚合物(PAPI):
CHO(CH:CHO),H
「H
CH:O(CHCHO),,H
n=0,1,2,3,·· CH
为了降低挥发性和毒性,可以制造异氰酸酯的加成物,如甲苯二异氰酸酯与二乙二醇的加环氧丙烷与三羟甲基丙烷的反应产物:
成物: CH2O(CH2-CHO)nH
H,C CH
CH, CH1CH2C-CH2O(CH2CHO)n2H
OCN NCOOCH:CH:OCCH2CH2OOCN
NCO
H CH1
H
甲苯二异氰酸酯与三羟甲基丙烷的加成物:CH:O(CH:CHO)n,H
CH:00CNH -NCO
CH
环氧丙烷与木糖醇的反应产物:
CH1-CH:-C-CH:OOCNH-
CH CH2O(CH,CHO)m,H
NCO CH3
CH CH:OOCNH
-NCO CHOCCH2CHO)nH
CH
CH CHO(CH,CHO)n,H
还可制成具有异氰酸酯基的三聚氰酸酯聚合物(常以甲苯二异氰酸酯10%~50%溶液)的1
形式应用: CH1
CH, CHO(CH,CHO)n,H
HD
CH
CH CH3
OCN NCO
(CH2) (CH2)NCO
CH2O(CH:CHO)n,H
OCN CH
用环氧乙烷代替部分环氧丙烷,在聚醚中引入伯羟基,可提高反应活性。多元胺起始剂有乙二胺、二亚乙基三胺等。如环氧乙烷与乙二胺反应可得到:
HO
NCO NCO
CH3 1
NCO NCO
H(OCHCH2)n (CH2CHO)n,H
CH
HD
CH CH3
9.1.2 多羟基化合物和聚合物NCH:CH2N
H(OCHCH2)n: (CH:CHO)m,H
1 CH
包括聚醚和聚酯两大类。CH
用于异氰酸酯制造的多羟基化合物主要是多元醇,如1,4—丁二醇和蓖麻油。多羟基聚合物
138 139
聚合物基复合材料
CH 第9章 聚觀酯树脂
CH: ①二月桂酸二丁基锡(DBTDL)DBTDL的生产由丁醇、碘、磷反应生成碘丁烧。碘丁烷
H(OCHCH2) (CH:CHO)n,H
NCH:CH,N (CH:CHO)n,H
丁基锡。
HCOCHCH:): 二月桂酸二丁基锡催化剂的毒性较大,操作时应注意劳动防护。空气中最高容许浓度为
CH CH
(2)聚酯 聚酯是由二元酸与多元醇在醇过量的条件下缩聚而得,二元酸有乙二酸、0.1mg/㎡',溶于多元醇和有机溶剂。
②辛酸亚锡(2—乙基已酸亚锡)2—乙基己酸与氢氧化钠反应生成2—乙基已酸钠,然后与氯
三羟甲基丙烷等,用于制造聚氨酯的聚酯分子量为400~6000,常用的是1000~3000。
化亚锡在惰性溶剂中加热进行复分解反应制得2—乙基已酸亚,原料2已已酸钠,然后与氧水和氯化亚锡物质的量比为1:0.516:0.5.反应过程中加入少量防老利—254,可提高2—乙
由己二酸与乙二醇制得的聚酯为:
(n+1)HOCH,CH,OH+nHOOC(CH2),COOH→ 基已酸亚锡的锡含量和稳定性。
辛酸亚锡溶于多元醇和大多数有机溶剂,不溶于醇和水。辛酸亚锡至少可存放12个月,但
H[OCH,CH,OOC(CH,),CO].OCH,CH,OH+2nH2O
容器必须密封,须储存于干燥处,防止高温以及过大的湿度。辛酸亚锡无毒性与腐蚀性,可用于制造医疗用品。
9.1.3 助剂 (2)权胺类催化剂 叔胺类催化剂对促进NCO/H,O反应特别有效,一般用于制备聚氨酯
聚氨醋胶黏剂制造中除异氰酸酯和多元醇基本原料外,添加各种助剂也是很重要的。助剂泡沫料。发泡型聚氨酯胶黏剂以及低温固化型、潮气固化型聚氨酯胶黏剂也采用。
可改进生产工艺,改善胶黏剂施胶工艺,提高产品质量以及扩大应用范围。叔胺类催化剂有4种类型:脂肪族类有三乙胺、二亚乙基三胺、二甲基十六胺和双二甲基
9.1.3.1 溶剂氨基乙基醚(NIAXA—1催化剂)等;脂环族胺类有三亚乙基二胺、N—乙基吗啡啉、N.N'—二
为了调节聚氨酯胶黏剂的黏度,便于工艺操作,在聚氨酯胶黏剂的制备过程或配制使用时,甲基环已胺等;醇胺类有三乙醇胺、甲基二乙醇胺、二甲基乙醇、乙醇胺等;芳香胺类有吡啶、
经常要采用溶剂,聚氨酯胶黏剂和涂料用的有机溶剂必须是“氨酯级溶剂”,基本上不含水、醇N,N'—二甲基吡啶等。主要品种介绍如下。
等活泼氢的化合物。“氨醋级溶剂”是以异氰酸酯当量为主要指标,即消耗1mol的—NCO所需①三亚乙基二胺 化学名为1,4—重氮双环[2,2,2]辛烷,结构式如下:
CH2-CH: 溶剂的质量(g),该值必须大于2500,低于2500以下者为不合格。因此,聚氨酯胶黏剂用的溶
剂纯度比一般工业品等级高。不-H0-N
CH:-CH: 聚氨酯胶黏剂采用的溶剂通常包括酮类(如甲乙酮、丙酮)、芳香烃(如甲苯)、二甲基甲
三亚乙基二胺是一种笼状化合物,两个氮原子上连接3个亚乙基,这个双环分子的结构非酰胺、四氢呋喃等,溶剂的选择可根据聚氨酯分子与溶剂的溶解原则,即溶度参数SP相近、极
常密集和对称。分子结构中的N原子无空间位置影响,一对空电子易于接近,因此三亚乙基二性相似以及溶剂本身的挥发速度等因素来确定。可采用混合溶剂来提高溶解性、调节挥发速率
胺对异氰酸酯基和活性氢化合物有极高的催化活性。适应不同粘接工艺的要求。
9.1.3.2 催化剂三亚乙基二胺在常温上为晶体,使用不方便,因此将三亚乙基二胺用一缩丙二醇配制成含
量为33%的溶液(牌号为Dabco—33—LV),其特点是黏度低,易于操作,同时保持了三亚乙基二
制备聚氨酯树脂中主要有3种反应需用催化剂:催化NCO/NCO(异氰酸酯二聚或三聚)、胺的催化能力。
NCO/OH(异氰酸酯与多元醇反应)、NCO/H2O(异氰酸酯与水反应)。制造聚氨酯树脂主要②三乙醇胺 醇胺类催化剂活性较三亚乙基二胺小,其优点在于能使反应物料的操作时间
用NCO/OH反应催化剂和NCO/H,O反应催化剂。延长,常和其他催化剂并用。另一特点是分子中具有羟基,与异酸酯反应后成为结构的一部分。
(1)有机锡类催化剂 有机锡类催化剂催化NCO/OH反应比催化NCO/H2O反应要强,聚三乙醇胺分子式为C,H15O3,分子量为149.19。外观为无色黏稠液体,在空气中会逐渐变
氨酯树脂制备时大多采用此类催化剂。有机锡类和胺类催化剂对异氰酸酯反应的相对活性黄。有吸湿性,溶于水、乙醇和氯仿,微溶于乙醚和苯。对皮肤和黏膜有刺激性,但不强。无
见表9-2。
强烈的氨味,使用方便。物性指标:相对密度(d)1.1242,熔点20~21℃,沸点360℃,黏
表9—2 有机锡类和胺类催化剂对异氰酸酯反应的相对活性度613.6mPa·s,闪点(开杯)90.55℃,折射率(n)1.4852。
催化剂 含量/%
NCO/OH反应的相对活性NCO/H2O反应的相对活性
③三乙胺 三乙胺也是第三胺催化剂,中等催化活性,一般都和其他催化剂并用。分子式
无 0.0
为C,HN,分子量为101.19。它是易挥发的无色液体,有强烈的氨味。物性指标:熔点
一
四甲基丁二胺 0.1
0.0
56.0 —115℃,沸点89.90℃,相对密度(d)0.7255,折射率(n)1.4003。能溶于乙醇和水。
三亚乙基二胺 1.6
0.1 130.0
9.1.3.3 扩链剂与交联剂
0.1 210.0
含羟基或含氨基的低分子量多官能团化合物与异氰酸酯共同使用时起扩链剂和交联剂的作二月桂酸二丁基锡
27.0
0.1 540.0
用。它们影响聚氨酯硬段和软段的关系,并直接影响聚氨酯产品的性能,所以配方中要用扩链辛酸亚锡
1.3
与交联剂。扩链剂能与过量异氰酸酯进行二次反应,生成脲基甲酸酯或缩二脲结构而成为交联
1.0
剂。扩链剂与交联剂有醇类和胺类,醇类有1,4—丁二醇,2.3—丁二醇、二甘醇、1,6—己二醇、甘
140 油、三羟甲基丙烷、山梨醇等。胺类有3.3—二氯—4.4'—二氨基二苯基甲烷(MOCA)以及
141
聚合物基复合材料
第9章 聚氨树脂
MOCA用甲醛改性制成的液体MOCA等。
过氧化氢,加人空间位阻除及芳族伸酸作抗氨防老剂,与亚磷酸酯、脾、硫税等化合物组成复
可以期)其本了结构中的数硬度,300的合成方法有乙块法和二氧了烯法两种,目前主要它
合用基g(3.5—二叔丁基4—羟基米)丙酸季成四醇酯(抗氧剂1010)是,数平基二米酸
乙炔法为主。
化钙成分于后中相利进行脱水,经减压蒸督后水分含量可低于0.1%6。1,4—丁三醇也可以同数须面树游其光稳定性效果更好,适合聚氨酯树脂应用的紫外线吸收剂者复合在一起加人聚
醚或聚酯多元醇混合一起脱水。33二面,小二氨基二苯基甲烷 该产品的商品名称为MOCA,其结构式如下:
事,位阻胺常用的牌号为Tinuvin292,是瑞士Ciba公司产品。将抗氧剂1010和紫外线吸收剂UV—327复合加人聚氨酯材料中,添加量为0.1%~0.5%,
经老化试验,其耐老化效果特别显著。
D
聚酯型聚氨酯弹性体耐水解稳定性较差,一般添加碳化二亚胺(—N—C—N—)之类水解稳定剂(Bayer公司生产的牌号为Stabaxol P和 StabiaxolD)。
MOCA的生产工艺如下,将邻氯苯胶与盐酸反应生成邻氯苯胺盐酸盐,然后滴加甲醛缩合9.1.3.5
填料与触变剂
成MOCA租晶,中和,蒸馏出过量的邻氯苯胺,水洗,乙醇重结晶,然后加工成粒状球体(1)填料 聚氨酯树脂组成中添加合适的填料主要是为了改进物理性能。加入填料能起补
作用,提高聚氨酯的力学性能,降低收缩应力和热应力,增强对热破坏的稳定性,降低热膨出售。
MOCA可溶解在丙酮、二甲基亚砜、二甲基甲酰胺、四氢呋喃等有机溶剂中。MOCA还可胀系数,另外还可改进树脂的黏度和降低成本。
溶于加热的聚醚多元醇中,有利于聚氨酯树脂配方的调节。MOCA强烈刺激呼吸道,50%死亡聚氨醋胶黏剂常用的填料有碳酸钙、滑石粉、分子筛(粉末)、陶土等。添加前的填料需经
率的照射剂量(LD2)为5000mg/kg。近年来有报道指出,MOCA是化学致癌物质,因此使用过脱水处理,以避免消耗掉部分异氰酸酯。须注意生成二氧化碳会导致树脂出现发泡现象,影
时要加强通风,尤其要避免吸入其蒸气,MOCA主要在聚氨酯弹性体和聚氨酯胶黏剂中作为扩响聚氨酯树脂的物性。
链剂。 ①碳酸钙 碳酸钙填料一般是粒径为2~20μm的粉末,有合成和天然产品。合成产品有沉
(3)三羟甲基丙烷 三羟甲基丙烷(TMP)是制备聚氨酯弹性体、聚氨酯胶黏剂以及涂料淀碳酸钙或轻质碳酸钙:天然产品由石灰石粉碎制得,也称重质碳酸钙或白垩。质量好的品种
时用的交联剂。从结构上看,TMP中的羟基为伯羟基,因此反应活性比甘油大。其化学结构式是方解石经粉碎并进行表面处理的产品。
如下: ②陶土 陶土的结构为Al,O3·2SiO2·2H:O,天然产品称高岭土或瓷土。粉碎后粒径在
HO-HO 0.2~10μm范围。粒径大的瓷土吸水性小,消光效果好;粒径小的瓷土1μm以下,可用于半光
CH-CH:-C-CH:-OH 涂料和内用涂料。
CH2-OH ③滑石粉 滑石粉的结构为3MgO·4SiO:·2H2O,其粒径在6μm以下。滑石粉(水合硅
三羟甲基丙烷生产工艺:由丁醛与甲醛于氢氧化钠溶液中进行缩合反应,经浓缩除盐,用酸镁)中和氧结合的镁原子夹在两个片状二氧化硅之间,形成层状结构。而且层与层之间通过
离子交换树脂脱皂,最后进行薄膜蒸发制得。TMP基本无毒,采用一般防护方法即可。在市场很弱的范德瓦耳斯力结合,因此纯的滑石粉很软,层间容易分离。滑石粉可用作弹性补强剂。
上出售的TMP采用烯烃薄膜复合铝箔材料进行包装,以防止吸水。TMP易溶于水、乙醇、丙(2)触变剂 为了使聚氨酯胶黏剂在施胶过程中能控制胶液的流动性,在胶黏剂组成内添
酮、环己酮、甘油以及二甲基甲酰胺,微溶于四氯化碳、乙醚、氯仿,不溶于脂肪烃、芳香烃。加触变剂,尤其是粘接皮革、纺织物或混凝土这类吸附较强的材料时,加入粒子极细的二氧化
(4)氢醌—二(β—羟乙基)醚 氢醌—二(β—羟乙基)醚简称HQEE,其结构式如下:硅(气相白炭黑)填料作为触变剂,可防止胶黏剂对这些材料的渗透,聚氨酯胶黏剂和密封胶
HOCH:CH:-0 O-CH:CH:OH
中加入适当的触变剂可调节胶黏剂的黏度,粘接垂直面基材时可防止胶料的下垂,提高滞留
特性。 它由对苯二酚和环氧乙烷合成,用作聚氨酯弹性体和聚氨酯胶黏剂的扩链剂,其制品的耐
热性能、硬度以及弹性高于一般使用的扩链剂。聚氨酯树脂中应用的触变剂主要是气相二氧化硅,其平均粒径为4~7μm,相应的比表面积
(5)1,6—己二醇 1.6—已二醇简称HD,其结构式如下:为50~380㎡/g。一般采用比面积为200㎡/g的产品。当需要更高的触变性时,则使用表面积
HO-CH,-CH2-CH2-CH2-CH2-CH2-OH 更大的气相二氧化硅。德国Degussa公司的气相二氧化硅产品的技术规格见表9—3。
它可作为聚氨酯树脂制备中的扩链剂,也可以同已二酸经缩合反应制成聚己二酸—己二醇
表9—3 气相二氧化硅产品的技术规格
酯,或与其他酸进行二元醇共聚,这种聚酯多元醇制得的聚氨酯制品其耐水、耐热、耐氧化以
牌号
Aerosil-300 Aerosil-380
Aerosil-CoK8 及机械强度等均能提高。
Aerosil-200
9.1.3.4 稳定剂BET表面积/(㎡2/g)
200±25 300±30
380±30 170±30
7 7
平均原始颗粒度/nm12
聚氨酯树脂也存在着老化问题,主要是热氧化、光老化以及水解,针对此问题须添加抗氧剂、光稳定剂、水解稳定剂等予以改进。
压实表观密度/(g/L)50
50
抗氧剂的作用是阻滞聚氨酯热氧化作用,阻止由氧诱发的聚合物的断链反应,并分解生成
(常规)
50 50
142 143
聚合物基复合材料
第9章 聚氨酯树脂 续表
是此反应为亲核加成的间接证据,这样,芳族异氧酸脂比脂防族异氧酸酯反应活性大。面含活Aerosil-200
Acrosil-300 Aerosil-380
Aeronil-Cokg 排氧化合物与异氰酸酯的反应速率取决于化合物的亲核性,按下屏系酸脂反
R,NH> RNH,>NH,>C,H,NH,>ROH>H,O> C,H,OH>RSH> RCOOH 牌号
120 120
120
(常压) <1.5
<1.5 <1.5
重要的反应如下。
含水量(105℃/2h)/%<1.5
(1)与醇、酚的反应 异氟酸酯与羟基化合物的反应由羟基化合物对异氧酸酯上碳原子的<2
<2.5
>
灼烧损失(1000℃/2h)/%<1
亲核进攻引起。接着羟基上的氢向氮原子转移,生成氨基甲经器,3.6~4.3
3.6~4.3 3.6~4.3
3.6~4.3 pH值(4%水悬浮体)
>99.8 >99.8
>99.8 82~86
R-N-C-0+R'OH
O
SiO2含量/% <0.05
<0.05 e
<0.05 14~18
HOR HOR
Al:O1含量/% <0.003
<0.003 <0.1
由于亲核性不同,酚与异氰酸酯的反应活性不及醇,一般在50~75℃下反应也很缓慢,需OR'
<0.003 FeO2含量/%
<0.03 <0.03
<0.03 <0.03
要加入催化剂。 Ti2O含量/%
<0.025 <0.025
不同醇与异氰酸酯的反应活性不同。伯醇与异氰酸酯的反应在室温下无催化剂即可进行。HCI含量/%
<0.025 <0.1
在同样条件下,仲醇的反应速率为伯醇的3/10,而叔醇仅为0.5%,三苯基甲醇甚至不起反应。■渣(45μm)/%
<0.05 <0.05
<0.05 <0.1
异氰酸酯上的亲电基增加碳原子上的正电荷,使反应活性增大。这样,便有以下的活性
9.1.3.6 其他助剂次序:
(1)偶联剂 为了改善聚氨酯胶黏剂对基材的粘接性,提高粘接强度和耐湿热性,可在胶O2N
NCO><NCO>H:C- -NCO>C,H2+,NCO
液中或底涂胶中加入0.5%~2%的有机硅或钛酸酯类偶联剂。常用的有机硅偶联剂有7—氨丙基实际上,4,4—二苯甲烷二异氰酸酯、甲苯二异氰酸酯比六亚甲基二异氰酸酯活性大。至于
三乙氧基硅烷(KH—550)、环氧丙氧基丙基三甲氧基硅烷(KH—560)、苯胺甲基三乙氧基硅甲苯二异氰酸酯的两种异构体,由于邻位—NCO受到的空间位阻较大,所以2,4—甲苯二异氰酸
烷等。 酯较2,6位的活泼。
(2)增黏剂 与聚氨酯胶黏剂组成中加入增黏剂可提高胶黏剂的初黏性和黏度,常用的增在二异氰酸酯中,当第一个—NCO与羟基化合物反应生成氨基甲酸酯后,未反应的—NCO
黏树脂有萜烯树脂、酚醛树脂、萜烯酚醛树脂、松香树脂、丙烯酸酯低聚物、苯乙烯低聚物。活性较小。利用这一点,工业上常使较活泼的第一个—NCO与羟基化合物反应生成加成物,即
(3)增塑剂 为了改进聚氨酯胶层的硬度,可加入少量增塑剂。但应在不损失黏合强度的封闭体。留下的—NCO待需扩展分子链或固化时起作用。
条件下增加柔韧性和伸长率。增塑剂有邻苯二甲酸二辛酯、二苯甲酸二乙二醇酯等。(2)与羧酸的反应 羧酸中也会有羟基,因此也能与异氰酸酯反应。其反应活性较伯醇与
(4)杀虫剂 聚酯型聚氨酯树脂易于受微生物侵袭,特别是在湿热、高湿环境下使用时更水低。羧酸与异氰酸酯反应先生成中间产物,中间产物的稳定性因羧酸和异氰酸酯的结构不同
易受微生物侵袭,因此,在聚氨酯树脂组分中常加入抗细菌、酵母或真菌的杀虫剂,常用的杀而异。脂肪族羧酸和脂肪族异氰酸酯反应先生成混合羧酸酐,然后分解产生酰胺,放出二氧
虫剂有8—羟基喹啉铜、三丁基氧化锡及其衍生物。化碳:
(5)着色剂 在聚氨酯树脂中添加着色剂可使之成为有色树脂。添加方法是将无机或有机0 0
1 颜料与多元醇混在一起制成糊状物,加到多元醇配方中。无机着色剂有二氧化钛、氧化铁、氧
RNCO+R'COOH→[RNHC-O-CR']→RNHCOR'+CO:↑
化铬、硫化镉、铝酸镁、炭黑等。有机颜料有偶氮/重氮系染料、酞菁及二噻嗪等。也有的厂家脂肪族羧酸或弱的芳香族羧酸与芳香族异氰酸酯反应:
如德国Goldschmid公司生产专供聚氨酯树脂着色用的色浆(牌号Tegocolor,有红、黄、绿、0
0 /
/
蓝、黑五种颜色),颜料含量为10%~20%,分散在多元醇中,羟值为56mg KOH/g,储存期ArNCO+R'COOH→[ArNHC-O-C-R']
为12个月。
0
0 0
0 0 0
11
1 /
1
9.2 聚氨酯的合成原理2[ArNHC-O-C-R']→[ArNHC-O-CNHAr]+R'COCR'
ArNHCONHAr+CO2
9.2.1 异氰酸酯的化学反应当温度升到160℃时,脲和酸酐又可进一步反应生成酰胺,同时放出二氧化碳:
00
11
异氰酸酯具有较高的反应活性,容易与含有活性氢的物质(醇、酚、酸、胺、水等)反应。
ArNHCONHAr+ R'COCR'→2ArNHCOR'+CO2
这个反应是由含氢化合物对异氰酸酯中的碳原子的亲核进攻引起的亲核加成:
(3)与水的反应 异氰酸酯与水反应经过不稳定的中间产物氨基甲酸生成胺和二氧化碳,
R-N=C-0+BH 胺又迅速与异氰酸酯反应生成取代脲:
eBH RNCO+H2O[RNHCOOH]→RNH2+CO2
异氰酸酯中R的电子接受体效应以及含氢化合物中B的电子给体效应都可加速此反应,这
RNH:+RNCORNHCONHR
144 145
聚合物基复合材料
2RNCO+H:0→RNHCONHR+CO2 醇的反应为例:
第9章 聚氨酶树脂
总的反应可写成:
OCN-(CH:)-NCO+HO(CH2),OH→OCN-(CH2)-NHCOO(CH2).OH 生成的二氧化展可作为气治来源。故此反应是制造泡沫塑料的基本反应。水与异氰酸面的
OCN-(CH2)。-NHCOO(CH2),OH+OCN-(CH2)(-NCo一
OCN-(CH2),-NHCOO(CH2),OOCNH(CH2)-NCO 2活世相当于所度 并新酸酯与酸的反应活性较与其他活性氧化合物为高,反应生成取代
最后生成:
胺的碱性越强,则活性越大。脂肪胺的活性比芳香胺大。EOCNH(CH2),NHCOO(CH2),03
用多官能团化合物(多元异氰酸酯或多元醇)可形成体型聚氨酯。按的强性题货,以上介绍的是初级反应,当异氯酸酯与反应物为等当量比时,主要进行
除上述反应之外,还可能发生诸如微量水与异氰酸酯的反应、次级反应以及异氰酸酯的自级反应。而当异氰酸醋过量时,初级反应产物可继续与异氰酸酯反应,即次级反应:
RNCO+-NHCOO-→RNHCONCOO- (脲基甲酸酯)
聚反应等副反应。
由于次级反应的存在,即使所用的两种原材料皆为二官能度,当二异氰酸酯过量时,仍可RNCO+-NHCO-→RNHCONCO
(酰脲) 能产生支化甚至交联反应。次线反应的进行与反应条件有关,通常在酸性条件下不利于次级反
RNCO+-NHCONH-→RNHCONCONH
(缩二脲) 应,故易形成线型产物,而在碱性条件下则有利于此反应,易形成分支或交联结构。
欲制造高分子量线型聚氨酯,必须严格控制反应条件,抑制副反应;另外,可通过副反应以上反应归纳见表9—4所列。
达到特定目的,如泡沫料的发泡、弹性体的硫化、漆膜的交联干燥等。表9—4 异氰酸酯的重要加成反应
反应产物 9.3 聚氨酯的制造工艺
与—NCO加成的基团初级反应
次级反应 早期制造聚氨酯采用熔融聚合。但这种方法的应用受到限制,生成的聚合物在熔融温度下
-NC-0-0-
-OH NH-COO
必须是热稳定性的。当聚氨酯熔融温度在225℃以上时,由于会发生热分解,不宜用熔融聚合,(氨基甲酸酯)
CO-NH
(脲基甲酸酯) 而应采用溶液聚合。
NC-O- 线型热塑性聚氨酯由接近等物质的量之比的二异氰酸酯与二元醇反应面得(NCO/OH约为
NH-CO-+CO: 1).分子量较大,可达30000~60000。采用低分子量二元醇,可以制得刚性的硬聚合物,而用
-COOH (酰脉)
C-ONH
(酰脲) 高分子量二元醇(分子量400~6000的聚酯或聚醚),则得到软的弹性合物,以六亚甲基二异氰
酸酯和丁二醇为基础的线型硬聚氨酯应用较多,它既可用溶液法,也可用熔融法制造。N-CO-NH-
NH: NH-CO-NH-
(取代脲) CO-NH-
9.3.1 熔融法 (缩二脲)
在反应釜中加入丁二醇,在氮气保护下加热到85~95℃,剧烈搅拌下,分小份慢慢加入六
除加成反应外,异氰酸酯的—C—N双键还可以自聚而成二聚体、三聚体:亚甲基二异氰酸酯。放热反应结束后,温度升到190~210℃,保持此温度至反应完成。然后,
CO 反应釜抽空以除去溶解的气体。用氮气将聚合物压出成带状,冷却,打碎。
8 N- (二聚异氰酸酯)
9.3.2 溶液法 R-NCO
在反应釜中加入氯代苯、二氯代苯和丁二醇混合溶剂。向加热到60~65℃的溶液中加入六
(三聚异氰酸酯) 亚甲基二异氰酸酯。保持沸腾4~5h,形成的聚合物以粉末状或絮状沉淀,过滤,在65℃下抽
空干燥到便得到线型热塑性聚氨酯。用高分子量二元醇制备聚氨酯与上述方法相似。高分子量聚酯或聚醚制得的软热塑性聚氨酯用于制造弹性体。而低分子二元醇制得的硬热
塑性聚氨酯用于制造塑料,可通过模压、注射、挤出、压延而成型。此外,还可制成纤维。
得到的二聚体不稳定、三聚体较稳定。欲得到高分子聚合物,则要在特殊引发剂存在并在另一类聚氨酯树脂是聚氨酯预聚体。这是分子量较小的聚氨酯,多数情况下,端基为
低温下反应。
—NCO,有时端基为—OH。根据NCO/OH不同,预聚体有以下几种:
9.2.2 聚氨酯的生成反应
NCO/OH<1
端基为-OH
多元异氰酸酯与羟基聚合物之间的反应为逐步加成聚合反应,没有副产物生成。
NCO/OH=1
端基为-NCO与-OH
线型聚氨酯由二元异氰酸酯与二羟基化合物或聚合物制得,以六亚甲基二异氰酸酯与丁二
1<NCO/OH≤2
端基为—NCO,无游离异氰酸酯
146 147
聚合物基复合材料
NCO/OH>2 端基为—NCO,有游离异氰酸酯
预聚体可用于二步法制造泡沫塑料、弹性体、漆与胶黏剂等。催化剂可加速聚合反应,可以采用叔胺催化剂或有机金属化合物,如前所述。
第10章 热塑性树脂基体
快,将液聚合中的把二甲基乙胶胺、二甲基亚是有效的溶剂,此外,还可以采用混合解如二甲基亚砜与4—甲基—2—戊酮或二氯甲烷、氯代苯与二氯代苯等。
10.1 聚乙烯
9.4 聚氨酯的应用
的性树脂,是世界产量最大、应用最广的合成树脂品种,品种繁多,分法各得,在能的通用热聚乙烯(PE)是以乙烯为单体,经多种工艺方法生成的一类具有多种结构和性能的通用热
9.4.1 聚氨酯泡沫塑料
153年,德国化学家齐格勒采用TiCl,—AIEt,为催化剂,低温低压聚合,制备了在密度聚乙烯。9.4.2
聚氨酯弹性体 10.1.1 合成
9.4.3 聚氨酯涂料
用乙烯为原料制备聚乙烯。根据聚乙烯合成压力大小,可分为低压法、中压法和高压法3种。
9.4.4 聚氨酯胶黏剂
高压法生产的聚乙烯是在1500~2000atm、180~200℃下,以氧作为引发剂,乙烯生成游离基,然后引发聚合成聚乙烯。它是分支较多的线型大分子,分子结构缺乏规整性,其分子
式为:
-CH:-CHECH2-CH2CH-
拓展阅读 (CH2),
(CH2),
HO
CH3
其分子量在25000左右,相对密度低,一般为0.910~0.925,所以叫低密度聚乙烯。
二实()
二 中压法生产的聚乙烯是在30~50atm或较高的压力下,用金属氧化物作催化剂,使乙烯在
庚烷、辛烷等溶剂中聚合成为线型聚乙烯。聚合反应是按离子型聚合机理进行的。一种是用分散于载体SiO2—Al2O3上的氧化铬为催化剂,在35~40atm、125~150℃下使乙烯聚合;另一种是以分散于Al2O3载体上的氧化铂为催化剂,在50~100atm下使乙烯在溶剂中聚合。前者制得的聚乙烯分子量为48000~50000,密度为0.95~0.97,结晶度为90%以上。
低压法是在1~5atm、60℃下,用齐格勒—纳塔催化剂,在烷烃溶剂中,使乙烯聚合,制得含很少支链的高密度聚乙烯。其分子量为70000~350000,密度约为0.94~0.96,结晶度为
85%~95%。 10.1.2 性能
活■
聚乙烯分子中不含有极性基团,因此介电性、化学稳定性好,吸水性低,另外还有其他各
种优异性能。 聚乙烯无毒、无味、是乳白色蜡状固体。相对密度在0.91~0.96,制成薄膜是透明的。
工业用的聚乙烯软化温度在108~132℃,这样低的熔点是由于分子链较柔顺和分子间作用力不大所致。聚乙烯使用温度不高,高压聚乙烯使用温度约在80℃,而低压聚乙烯在无负载情况下,长期使用温度不超过121℃。然而在受力情况下,即使很小负载,它的变形温度也很低。
聚乙烯的耐寒性很好。分子量越高,支化越多,脆化点越低。聚乙烯的强度主要由结晶时分子紧密堆砌程度所提供。分子量的大小对它的性能有很大影
1atm≈105Pa。
149
聚合物基复合材料
第10章 热塑性树脂基体答,它龄力学在能以设在国大和度上来决于乘乙烯的分于里和文化度,强度不商,国
低,弹性模量小,容易蠕变和应力松弛。评些数量小,在是合势,它的绝涂性比其他任何分电物质都好,可以作超商频地维材,
聚丙烯 10.2
聚丙烯(PP)是由丙烯(CH2—CH—CH)合成的,而丙烯来源于石油裂化气、天然气乙的的电指的系,密度高的介电数也高,道落时密度降低,介电常数也低,此外,与是
烯在氧化时会产生羰基,使其介电损耗有所提高。
面广泛,价格低廉,生产工艺简单,产品性能良好,因此发展迅速。合成了高分子量、高熔点的结晶聚丙烯,1957年实现了大规模工业C,作催化剂
化时会产生乙势不受和硫酸和检硝酸的侵蚀,盐酸、氢氟酸、磷酸、甲酸、醋酸、、胺类、过氧化氢、氢氧化钠、氢氧化钾等,对聚乙烯均无化学作用。
乙烯,聚乙烯在SO2存在下氯化时,SO2和Cl2都可进入聚乙烯。其反应大致如下:、过氧化液时酸在鉴温下对聚乙烯能起缓慢破坏作用·而在90~100℃时,则能迅速破坏采
10.2.1 合成
原料丙烯在溶剂中采用齐格勒—纳塔催化剂,在常温~80℃、3~10MPa压力下聚合,得到CH:
CH CH:
CH
CH2 立体规整性很强的聚丙烯。
CH: CH: CH:+Cl2+SO2→CH CH
nCH1-CH→ECH2-CH]。 Cl
SO2CI CH2
CH2 聚乙烯能溶于热苯中,溶度参数相近的低分子物质只能使聚乙烯溶胀。聚乙烯是非极性囊
其等规度可达90%~95%。合物,胶黏剂胶接及印刷都困难,对其表面进行处理,可提高胶接和印刷的性能。
聚乙烯受到空气中的氧、紫外线辐射,其性能指标会降低。长时间氧化,特别在高温情况10.2.2 结构与性能
下,会使聚乙烯大分子降解,并析出CO2和H2O,最后生成脂肪酸和蜡状产物。所以一般聚乙聚丙烯是头—尾相接的线型结构,根据其侧链甲基(—CH3)的空间排列,有以下3种不同
烯造粒过程中加入抗氧剂,加入量大约为0.05%。的形式。
聚乙烯受到高能辐射时,一方面,分子间形成碳碳键而交联;另一方面,长时间照射会变全同立构聚丙烯:主链上的甲基全都排列在分子链的一侧,由于位阻效应,它的构型呈主
色,有空气存在时,还会发生表面氧化,也会引起聚乙烯降解;两种作用结果使聚乙烯性能变体螺旋状的结晶,等规度高达90%~95%。
劣。在氧存在的情况下,低辐射剂量长期照射的聚乙烯薄膜可能会引起严重的降解,而高辐射CH
剂量短期较厚的聚乙烯试样,其降解可能性不大。聚乙烯中加人炭黑可以提高其制品的耐辐射性。
H H
H
聚乙烯制品在受应力状态下或者与某种液体接触时产生龟裂,这种现象称为环境应力开裂,间同立构聚丙烯:主链上的甲基是交替地向主链的两个方向排列。
特别是分子量小时这种倾向更显著。在各种表面活性剂、矿物油、动植物油、酯类增塑剂、强CH
CH1
H H
碱、醇存在时,聚乙烯制品常产生这样的环境应力开裂。-CH:-C-CH2-C-CH2-C-CH2-C-
水几乎不能透过聚乙烯薄膜,但CO2、有机溶剂、香料等的透过率相当大,所以作为食品H
CH3 H
CH1
及其他包装材料使用时要特别注意。高密度聚乙烯比低密度聚乙烯透湿度和气体透过率小。无规立构聚丙烯:聚丙烯主链上的甲基是任意而无序排列的,是无定形的。
在通常情况下,聚乙烯是在熔融的状态下进行加工,如注射、挤出、吹塑或压制成型。CH3
CH3 CH1
H
10.1.3 用途 -CH2-C-CH:-C-CH2-C-CH2-C-
H H CH3
H
聚乙烯的介电性能良好,不易渗透水汽,因而能用于制造电话、信号装置、远距离操纵系
工业用聚丙烯具有高度空间规整性,主要包含全同立构的大分子链(85%~95%),有高度
统、高频装置和电气工程中的电线绝缘和电缆绝缘,以及水底电缆的绝缘、地下电缆外护套等。
的结晶性,同时含有少量非结晶的无规立构和低结晶的全同立构、间同立构及无规立构的嵌段
聚乙烯广泛用于制备各种管材,例如城市室外给排水管路网,牧场、农村、工厂的给排水,
的大分子链,熔点在170~175℃范围内,高聚物的立体规整性愈高,熔点也愈高。聚丙烯的Tg
室内自来水管和排水管,泵站的管道和室内灌溉网。交联聚乙烯管子可用于热水给水系统。热
与结构和结晶度有关,它明显地依赖于聚合物中非品态部分含量。90%以上全同立构大分子的
塑性塑料管用于输送天然气是一项比较新的使用领域。还可以用于发泡保温材料。
聚丙烯,结晶度为67%~70%时,Tg为—35℃。
大量聚乙烯可制造有广泛用途的薄膜,聚乙烯主要用于生产农业育秧膜、大棚膜、地膜、
工业用聚丙烯的分子量一般在150000~700000的范围内,随不同产品要求而异,与其他高
食品包装膜等薄膜。
聚物比较,聚丙烯分子量的分布较宽,宽分子量分布的聚合物的流变性对制品的性能有很大影
聚乙烯可以注射成制件或用压制而成板材,以及用冲压法或按样板弯曲的方法制成各种零
响,因此如果缩小其分子量分布,则制品的机械强度可明显改善。
件、家庭用品、玩具、化工和电气零件。高密度聚乙烯用辊压或火焰喷涂,或静电喷涂,可以
聚丙烯的各种性能与聚乙烯非常相似,它是塑料当中相对密度最小的一种(0.89~0.92),
作为化工设备中容器的耐腐蚀涂层,还可以代替钢和不锈钢制造阀门、衬套、管道等。其耐磨
与聚乙烯相比软化温度显著提高,全同立构聚丙烯的熔点170℃,拉伸强度、刚性、弯曲强度都
性好,可制小载荷的齿轮、轴承、冷冻仓库包装容器、油桶、周转箱、软瓶、牛奶瓶、盆子、
很高,在适当拉伸条件下处理,拉伸强度、刚性和冲击强度都提高,特别是耐弯曲疲劳性明显
瓶盖、冰箱盘子、盒等。
改善,比聚乙烯成品的透明性及表面光泽都好,成型时收缩率小,外观和尺寸精度都好。
150
151
聚合物基复合材料
第10章 热塑性树脂基体和5选与6称、自、请石的、或有行速复合,其力学性能有很大的改变,有曲性
②以乙烯为原料制备氯乙烯 以乙烯为原料制备氯乙烯的一般原则,必须制备对称的二氧是有的大变化,方要积、有在及的地性他,因为它用批律比较经,所以在电器工业上区
乙烷, 然后其热解脱氯化氢,即
CH2-CH2+Ch→CHCI-CH2CI
CH2CI-CH2CICH2-CHCI+HCI 它的副产物HCl,再与氧、乙烯混合,反应也生成二氯乙烷,这种方法叫做二步氧氯化法,
好的分电的度域性能良好,无论在常温或校高温度(70—100)条件下,其减及是目前较经济和较科学的方法。反应式如下
2CH2-CH2+4HC1+O2→2CH2Cl-CH2C1+2H:O 提党为社能良好,但易受发烟硝能及发烟硫酸的腐蚀;对水特别稳定,24h的要水
另外,还有一种方法叫混合烯快法,其特点是把石脑油或原油裂解所得的混合气体,不经
于0.0张和液体的透过性最小:聚丙烯可以在开水中煮,在135℃、100h的蒸汽中消毒也不
分离直接导人反应器,乙烯和乙炔用氯气处理之后转变为二氧乙能,然后二的之位钱解,不经小于0.01%。
乙烯和氯化氢,而氯化氢与乙炔加成,得到氯乙烯。制造氯乙烯的方法还有很多,就不在这里会被破坏。
芳香族碳氢化合物和氯代烃在80℃以上能溶解聚丙烯,在常温下溶胀。一介绍了。
(2)聚合反应 氯乙烯聚合的方法较多,一般采用悬浮聚合与乳液聚合,悬浮聚合是将氯方在丙烯主链上包含有叔碳原子,易受到氧、臭氧攻击而降解,因而聚丙烯耐氧化性差,
乙烯单体在引发剂和分散剂存在下,悬浮在水相中,在激烈的搅拌下进行聚合,粒子直径较大,防老化同题十分重要。由于聚丙烯的加工温度较高,因此在加热过程中,加入抗氧剂是必要的。
在50~100μm范围内。这种方法占聚氧乙烯总产量的95%。乳液聚合是将单体氢乙烯在乳化剂聚丙烯也容易在紫外线下老化,所以还要加入炭黑和有机的紫外线吸收剂。
作用和搅拌下,很好地分散在水相中成为乳浊液,加入水溶性的引发剂,在50℃左右进行聚合。乘丙烯熔融温度较高,熔融范围窄,而熔体黏度比较低,是一种比较容易加工的树脂。表
乳液聚合的树脂颗粒较细,粒子直径在0.5~1μm范围内。这种方法得到的树脂,主要用作糊观黏度随剪切速率增加而迅速降低。聚丙烯的熔体黏度对温度的敏感性不强。聚丙烯是熔体强
状树脂。 度较低,熔体是假塑性流体,其非牛顿性比聚乙烯更显著。
其他聚合方法如溶液聚合得到的聚氯乙烯作涂料和胶黏剂使用,本体聚合得到的纯度高,聚丙烯的透明性也比聚乙烯好,但是这一点还必须和优越的机械强度、耐热性统一考虑,
热稳定性和电性能好,适合作电绝缘材料。
作为包装材料才有好的效果,特别是为了增加透明度,成核剂的加入是有效的,加入成核剂促10.3.2 结构与性能
进生成极其微细的球晶,使透明度变好,韧性和在低温下耐冲击性能提高。聚丙烯薄膜的气体透过率比高密度聚乙烯大,所以作为化妆品、食品包装材料时必须注意。
聚氯乙烯是无定形结构的热塑性塑料,主链结构单元是“头—尾”相接的链接方式。
-CH:-CH-CH2-CH-CH2-CH- 10.2.3 用途
C1 d
CI
聚丙烯主要用于制备管材、容器、汽车及电器电子零部件、纤维及其织物和薄膜等制品,也有少量“尾—尾”(头一头)键接形式,由于侧基氯(CI)的影响,聚氯乙烯大分于实际是
尤其引人注目的是聚丙烯塑料经玻璃纤维等增强,制备高性能的复合材料制品。一个极性分子,由于极性基的影响,氯乙烯分子间力较大,不易形成结晶。因为是非晶态结构,
聚丙烯的机械强度与尼龙差不多,特别在耐酸耐碱的化学装置与衬里材料方面大量使用,所以聚氯乙烯没有明显的熔点,玻璃化转变温度在80℃左右。
如法兰、接头、泵叶轮、阀门配件、电缆被覆材料等。聚丙烯用于汽车内外装件及发动机机罩聚氯乙烯的相对密度约1.4,是白色粉末,耐水性、耐酸性、难燃性、电绝缘性好,除此之
内部件,此外,还用于无纺布、各种管材、医疗器械、家具、玩具、餐具、文体用品和日用品。外还耐很多溶剂,聚合度低或与多种成分共聚物容易溶解。
一般聚氯乙烯树脂在64~85℃软化,120~150℃有塑性,170℃以上熔融,190℃以上分解10.3 聚氯乙烯
放出氯化氢,聚合物颜色发生变化(黄→红→棕→黑),同时开始分解,遇有能产生游离基的物
质,如过氧化物,以及某些金属,如锌、铁等存在,即使痕量存在,聚氯乙烯也会激烈分解。
聚氯乙烯(PVC)是20世纪30年代出现的品种,产量占树脂第二位,由于性能好,价格适合加工的温度是150~180℃。
低廉,原料来源广,受到普遍重视。
氯乙烯的均聚物占整个聚氯乙烯的90%,聚氯乙烯的共聚物,主要是与醋酸乙烯的共聚物,
10.3.1 合成 其加工的温度范围变宽,加工流动性、表面光泽等方面得到显著改善。共聚的醋酸乙烯量增多,
(1)氯乙烯单体的制备聚合物的强度、软化点都下降。聚合度的大小对材料的性能与应用都有影响。当聚合度高于800
时,聚氯乙烯的强度基本不变,而加工变差了,常用的聚氯乙烯的聚合度在800~1200之间。
①以乙炔为原料制备氯乙烯 乙炔与氯化氢加成反应,是制备氯乙烯的最简单的方法。化
聚氯乙烯化学稳定性好,氧和臭氧不能明显地浸蚀聚氯乙烯,但某些氧化物会对它有破坏
学反应式如下
作用,例如浓的高锰酸钾溶液使聚氯乙烯表面受浸蚀,而过氧化氢影响不大。室温下,除了浓
CaC2+2H2O→CH=CH+Ca(OH)2 硫酸(90%以上)和浓硝酸(50%以上)以外,聚氯乙烯耐酸、碱性良好,但在60℃以上耐强
CH=CH+HCI→CH2-CHCI
酸性下降。
这种方法是以电石开始,电石乙炔法工艺虽然简单,但耗能大,“三废”多,成本高,属于
聚氯乙烯耐大多数油类、脂肪烃和醇类的浸蚀,但不耐芳烃、酮类、脂类和环醚。环己酮、
限制和改造的工艺范畴。
四氢呋喃、二氯乙烷等是聚氯乙烯的溶剂。
152 153
聚合物基复合材料
第10章 热塑性树脂基体
吉七国·心主的的必愈期理,世3评,都的,版编·范·篇编本作
10.3.3 用途
用额城极广,从建筑材料、化工管道到儿童玩具,从工农业所用机械零件到目能造成裂纹或开裂。
廣苯乙烯使用温度一般在—30~80℃,热性能与分子量的大小,低分子量聚合物的含量和
常生活用品,按其加工方法划分其用途。
碱酸收据、排气塔、储槽、泵、风机及其代替船的防腐化工设备,或者用于钢管衬里层。其截少内应力,改进机械强度,提高热变形温度,聚苯乙烯的热导率做在65—8C下退大,可
收据,排气的可做建筑材料,如地下水道、废水管、阻燃的门、地板和墙壁材料。乙此小、定向度和含杂质的量有关。分子量高的机械强度高,分子量在方制造方法、分子雇的
始由于绝缘性的主要应用领域,广泛用作椅子、防雨布、各种箱子、沙发、衣服、办公用品和次于聚四氟乙烯。聚苯乙烯常采用注射成型方法,软化点低,菇融温度范倒,高级下电性能仅
家具等。 好,即使是比较复杂的零件,也可以充满模具,其次采用的方法是挤出成型。
10.4 聚苯乙烯 10.4.3 用途
聚苯乙烯(PS)是1930年以后发展起来的,尤其是1946年以后发展更快。聚苯乙烯的化、责体等;二是包装容器,如食品包装容器;三是日用杂品,如梳子、盒、牙刷柄、圆珠笔
聚苯乙烯的主要用途在3个方面:一是电气用品,如电视机、录音机以及各种电器仪表零学稳定性、电性能都很优良,而且容易成型,但主要缺点是机械强度不高,质硬而脆,耐热
性低。 以及儿童玩具。
此外,聚苯乙烯能制成质轻的泡沫塑料,能防震、保温,用在建筑工业上。也在电冰箱、10.4.1 合成
火车、船、飞机上用来隔声、隔热。还可做耐腐蚀设备,如储酸槽等。苯乙烯单体的原料是苯和乙烯,经过乙基苯催化脱氢得到苯乙烯单体。
CH2-CH: CH:-CH
10.5 ABS
+CH2-CH;→
Δ
苯乙烯单体在热、光、催化剂作用下很容易聚合成无色透明的聚苯乙烯树脂。10.5.1 合成
nCH:-CH→ {CH2-CH ABS树脂的制造方法有3种,即共混法、接枝法和接枝共混法。
(1)共混法 把AS树脂与NBR在加热情况下,用研磨法进行机械混合而成或者把树脂与
橡胶制成胶乳状再混合。这种共混型ABS树脂,使用的橡胶交联度对树脂和橡胶的相容性有很-1500~3000
大影响,交联度大则冲击强度大,因此在制造NBR时或塑炼时加入少量交联剂以提高其交苯乙烯的聚合方法很多,主要有本体聚合、悬浮聚合和乳液聚合等几种。
联度。
用本体聚合制成的聚苯乙烯电绝缘性极好,耐化学腐蚀性优良,可用压制、挤出法成型各(2)接枝法 ABS树脂是用聚丁二烯胶乳、苯乙烯和丙烯腈单体聚合制得的。但是,这种
种电绝缘用制品,因其透明性好,用在一些工业品和日用品上。类型的树脂,实际上包含接枝共聚物(以聚丁二烯为主链,以苯乙烯、丙烯腈共聚物为支链),
悬浮法生产聚苯乙烯,在聚合时要加入分散剂和引发剂。而采用高温加压悬浮聚合时,可苯乙烯与丙烯腈的无规共聚物(AS树脂)以及未接枝的游离聚丁二烯3种主要成分构成。
以不用引发剂,使生产效率提高,质量提高。
接枝型ABS树脂,除了乳液法之外,还可用本体或悬浮聚合方法或者本体—悬浮聚合法
乳液法生产聚苯乙烯通常用于制聚苯乙烯胶乳,作为涂料。采用这种方法容易控制反应温制得。
度,聚合速率也快,但乳化剂不易除去。
(3)接枝—共混法 这种方法是用接枝法制得的ABS树脂胶液与另外的AS树脂胶乳共混的
10.4.2 结构与性能方法。
聚苯乙烯的分子主链是饱和烃,侧基是苯基。由于聚合方法不同,含有一定的支链,聚合性能
10.5.2
温度越高,支链越多,由于分子结构不规整,苯基的位阻效应使分子链内旋转困难,因而大分
ABS树脂,因其制法,树脂的组成和分子量,橡胶的种类、组成、粒子半径、交联度、接
子刚性很大,不易结晶,因而,聚苯乙烯是典型的非晶态线型高分子化合物,质脆且透明。
枝率,树脂与橡胶的比例不同,性质会有很大的不同。以下介绍普通ABS树脂的基本性质。
聚苯乙烯是无色透明的材料,因此着色也很容易。它无味、刚硬、密度小,光稳定性和耐
ABS树脂是一种综合性能较好的工程塑料,外观微黄、不透明,并且无毒无味,分子量约
候性不如丙烯酸树脂,长期存放或日光照射时间长了,容易发黄,主要是反应杂质起促进作用
10000。拉伸强度和模量一般,但是具有优异的耐冲击强度,特别是低温下有很高的冲击强度,
所致,但是,抵抗放射线能力在塑料中最强。
而且热变形温度高。此外,电性能、耐化学药品性、耐油性好,还有加工适应性广,可以注射
溶解于许多溶度参数(δ=0.91)相近的溶剂中,如苯、甲苯、四氯化碳、氯仿、酮类(丙
成型、挤出成型、真空成型、吹塑成型等。尺寸稳定性好、耐蠕变、耐应力开裂,制品表面光
154 155








